Comments (16)
 chhylp123
commented on May 30, 2024
chhylp123
commented on May 30, 2024
Thanks so much for point that. It seems that the mis-assemblies are caused by the our new purge_dup. I will expose its parameters to users today, and let you know when it is available.
from hifiasm.
 lh3
commented on May 30, 2024
lh3
commented on May 30, 2024
v0.5 fixes one misassembly on our data, but generally hifiasm does produce large misassemblies occasionally. @chhylp123 will expose some purge_dups parameters, which may help. We are also thinking about the possibility to integrate some bogart heuristics in future.
from hifiasm.
chhylp123
commented on May 30, 2024
I have exposed three purge_dups parameters: '-l', '-s', '-O'. I guess it may help for fixing assemblies. Please use the latest commit with version 0.5-dirty-r247 (hash: 7f6725e). Looking forward to your results @HenrivdGeest .
from hifiasm.
 HenrivdGeest
commented on May 30, 2024
HenrivdGeest
commented on May 30, 2024
Thanks, I will start a sweep asap. Is their a way to re-use the error corrected reads? With some parameter testing, it can be faster to run that step once.
from hifiasm.
lh3
commented on May 30, 2024
If you specify the same "-o prefix" option, hifiasm will reuse "prefix.*.bin" files and skip error correction and overlapping. Note that older assembly graphs will be overwritten if you do this.
By the way, what is the heterozygosity of your genome? v0.5 outputs multiple k-mer histograms in stderr. Is it possible to show us the first histogram? Thanks.
from hifiasm.
HenrivdGeest
commented on May 30, 2024
I am running the loop now. The first kmer plot:

I also ran genomescope on kmer=21 on the hifiasm error corrected reads ( on more data, so the coverage peaks are at different numbers) before:
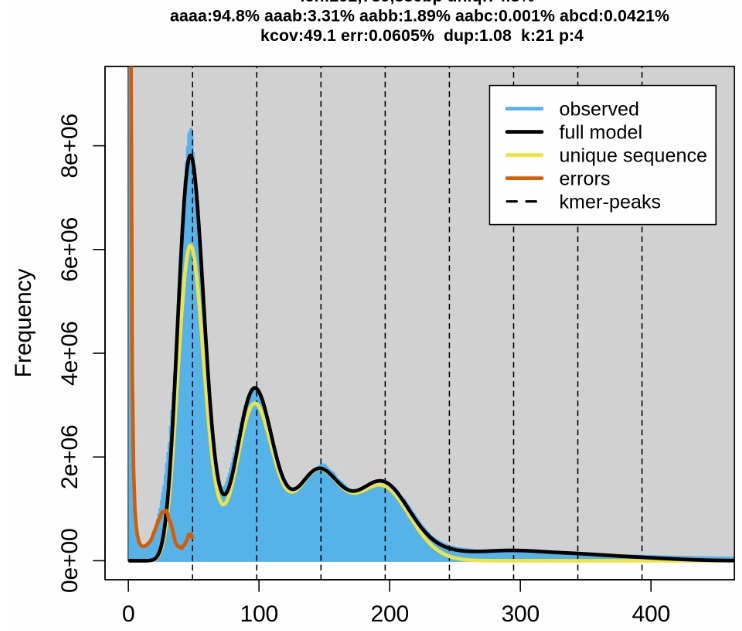
Here you see clearly 4 peaks. The hifiasm just 2, or barely 3, but that kmer is at 51, meaning that 2 haplotypes might already get completely separated at that size?
from hifiasm.
lh3
commented on May 30, 2024
Thanks a lot! Is this public data? We have been mostly using animal data for testing. The heterozygosity is much lower in comparison to some plant genomes. While redwood heterozygosity is high (higher than your genome) and its data is public, it takes several days to assemble, making testing very difficult. If your data is public or can be shared with us privately, it would be ideal for development purposes. Some hifiasm parameters are tuned for low heterozygosity, we may have a lot of room for improvement.
Also, what is the preferred output for your genome? Is it one primary assembly representing one haplotype?
from hifiasm.
HenrivdGeest
commented on May 30, 2024
I made 18 assemblies, all combinations of -l (0,1,2) -s (0,75,0,90) and -O (1,5,10). But all of them show exactly 2 misassemblies. I measured my version 0.3 the same, and that one has none. The n50's of 0.3 and the "v0.5 -l0,-s0.75 -l1" version are almost equal:

@lh3; My haploid genome size is around 400-600Mb, I was never able to collapse my assemblies to lower than 800Mb with purge_haplotigs, so I think some haplotypes are very heterozygous, and some not so, and can be easely collapsed. (hence the reason why I also never get a 2Gb genome fasta). Our plant is called a 'segmental allopolyploid' (so the other parts are autopolyploid).
Regarding what I want as output, in this case I want to have the unitigs, as much separating the haplotypes as possible. (I know all my previous remarks are about the contigs). The unitigs are upto 1.6Gb in size, but still contain collapsed haplotypes, probably because there are no snps to be found between alleles. This data is confidential, and can therefore, unfortenately, not be shared.
from hifiasm.
chhylp123
commented on May 30, 2024
I see. Thanks a lot. Let me check what's the difference between v0.3 and v0.5...
BTW, could you please run v0.5 on the bin files generated by v0.3?
from hifiasm.
chhylp123
commented on May 30, 2024
I think you can have try to further increase -s and -O. I guess even '-s 0.99' is fine for hifiasm. Actually '-s 1' doesn't mean hifiasm only purges exactly same haplotigs, it still allows differences.
from hifiasm.
HenrivdGeest
commented on May 30, 2024
I tried increasing -s to 0.999 and -O to 75, but that did not made any difference in the output regarding the misassembly. I tried running v05 on the v03 bin files, but for v03 I only have a *reverse.bin, and that alone doesn't seem to work, I think it starts assembling from scratch.
from hifiasm.
chhylp123
commented on May 30, 2024
Thanks a lot, I will expose another option to users. I believe that can avoid these two mis-assemblies.
from hifiasm.
HenrivdGeest
commented on May 30, 2024
BY any change any luck on these new options?
from hifiasm.
chhylp123
commented on May 30, 2024
Oh, I'm so sorry I forget that : ( . I will expose it this day.
from hifiasm.
chhylp123
commented on May 30, 2024
Please wait me one more day, I will fix it soon. Thanks a lot : )
from hifiasm.
chhylp123
commented on May 30, 2024
I have exposed '-u' to disable post-joining (0.7-dirty-r256). Hope it is helpful. I'm so sorry for the deay : (.
from hifiasm.
Related Issues (20)
- issue in generating hap1 and hap2 asm files
- larger assembly size than kmer estimation genome size HOT 1
- larger assembly size than kmer estimation genome size HOT 2
- Why more contigs always present in haplotype 1 than haplotype 2? HOT 2
- overlap parameter HOT 2
- How do you assemble chromosomes X and Y? HOT 3
- Add Options for Pore-C Data HOT 1
- Output interpretation with HiFi+ONT+HiC with inbred samples + `-l0` HOT 1
- low BUSCO scores HOT 1
- Mitigate Overlapping Sequence Assignments in Haplotypes HOT 3
- Help!!! Segmentation fault (core dumped) HOT 1
- Question about the depth of ONT ultra-long reads HOT 1
- Homotetraploid, super-large genome, with different parameters, the size of p_utg varies greatly? HOT 1
- setting K parameter in yak HOT 2
- how to make the correct genome size estimation for allotetraploid species? HOT 2
- Possible missing one haplotype in human assemblies HOT 2
- No haploid.gfa files output in trio-binning mode HOT 3
- Hifi + Hi-c + ONT assembly fails
- In Trio-binning, always more on hap1 despite (almost) same sequences for paternal and maternal
- discontinuous assembly with shorter pacbio hifi reads but high coverage HOT 2
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.
from hifiasm.