nolanlab / scaffold Goto Github PK
View Code? Open in Web Editor NEWscaffold
scaffold



















































Hi,
I succesfully installed scaffold on my windows 7 computer but it is impossible to run the script since when I choose a fcs file from a fcs file folder, an error message appears. Can it be a R3.4.1-R studio incompatibility with the script?

Hello,
I'm not able to find any kind of guide to run/deploy scaffold within a Shiny server to provide groups of users with an entry point on a managed environment.
Am I overlooking this or is scaffold intended to be single user only running from a development environment?
Thanks,
Martin
Hello,
I am struggling to get started with using scaffold in R. I have been able to load all the packages but when i get to loading my files nothing happens. I get the NULL response in R studio and I cannot choose a dataset or anything on the page in the browser.
Can you help?
Nyari
I downloaded a test fcs file "Blood_129.fcs" from Cytobank reports of Nolan Lab,
which is associated with a 2015 science paper
put it into a folder called "Cytof trial data" (as the working directory)
> scaffold.run()
I was prompted to select a file, as expected, so I chose to "Open" "Blood_129.fcs",
then I selected the "Run clustering" tab, and "representative FCS file" ("Blood_129.fcs", the only file),
selected the markers:
"CD45.2", "Ly6G", "CD11c", "IgD", "F480"
(all the other options as the default"),
and hit start clustering,
Data processing seemed to be completed smoothly:
Data processing is complete!
Clustering completed with markers CD45.2 Ly6G CD11c IgD F480
Files analyzed:
Blood_129.fcs
but when I checked the working directory, there were no expected newly created files:
my-fcs-file.clustered.txt
your-fcs-file.clustered.all_events.RData:
in the R studio,
> scaffold.run()
Listening on http://127.0.0.1:6077
NULL
Warning in parallel::mclapply(files.list, mc.cores = num.cores, mc.preschedule = FALSE, :
1 function calls resulted in an error
I have no idea what might go wrong, any suggestion would be welcomed.
Here is my R version and session information:
> R.version
_
platform x86_64-apple-darwin15.6.0
arch x86_64
os darwin15.6.0
system x86_64, darwin15.6.0
status
major 3
minor 4.0
year 2017
month 04
day 21
svn rev 72570
language R
version.string R version 3.4.0 (2017-04-21)
nickname You Stupid Darkness
> sessionInfo()
R version 3.4.0 (2017-04-21)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] shiny_1.1.0 scaffold_0.1
loaded via a namespace (and not attached):
[1] bitops_1.0-6 matrixStats_0.54.0 bit64_0.9-7
[4] RColorBrewer_1.1-2 GenomeInfoDb_1.12.3 tools_3.4.0
[7] backports_1.1.2 R6_2.2.2 rpart_4.1-13
[10] Hmisc_4.1-1 DBI_1.0.0 lazyeval_0.2.1
[13] BiocGenerics_0.22.1 colorspace_1.3-2 nnet_7.3-12
[16] tidyselect_0.2.4 gridExtra_2.3 DESeq2_1.16.1
[19] bit_1.1-14 compiler_3.4.0 graph_1.54.0
[22] Biobase_2.36.2 htmlTable_1.12 DelayedArray_0.2.7
[25] rtracklayer_1.36.6 scales_0.5.0 checkmate_1.8.5
[28] DEoptimR_1.0-8 mvtnorm_1.0-8 robustbase_0.93-1.1
[31] genefilter_1.58.1 stringr_1.3.1 digest_0.6.15
[34] Rsamtools_1.28.0 foreign_0.8-71 XVector_0.16.0
[37] rrcov_1.4-4 base64enc_0.1-3 pkgconfig_2.0.1
[40] htmltools_0.3.6 htmlwidgets_1.2 rlang_0.2.1
[43] rstudioapi_0.7 flowCore_1.42.3 RSQLite_2.1.1
[46] bindr_0.1.1 jsonlite_1.5 BiocParallel_1.10.1
[49] acepack_1.4.1 dplyr_0.7.6 RCurl_1.95-4.11
[52] magrittr_1.5 GenomeInfoDbData_0.99.0 Formula_1.2-3
[55] Matrix_1.2-14 Rcpp_0.12.18 munsell_0.5.0
[58] S4Vectors_0.14.7 stringi_1.2.4 SummarizedExperiment_1.6.5
[61] zlibbioc_1.22.0 plyr_1.8.4 grid_3.4.0
[64] blob_1.1.1 promises_1.0.1 parallel_3.4.0
[67] crayon_1.3.4 lattice_0.20-35 Biostrings_2.44.2
[70] splines_3.4.0 GenomicFeatures_1.28.5 annotate_1.54.0
[73] locfit_1.5-9.1 knitr_1.20 pillar_1.3.0
[76] igraph_1.2.2 GenomicRanges_1.28.6 corpcor_1.6.9
[79] geneplotter_1.54.0 biomaRt_2.32.1 stats4_3.4.0
[82] XML_3.98-1.13 glue_1.3.0 latticeExtra_0.6-28
[85] data.table_1.11.4 httpuv_1.4.5 gtable_0.2.0
[88] purrr_0.2.5 reshape_0.8.7 assertthat_0.2.0
[91] ggplot2_3.0.0 mime_0.5 xtable_1.8-2
[94] later_0.7.3 survival_2.42-6 pcaPP_1.9-73
[97] tibble_1.4.2 GenomicAlignments_1.12.2 AnnotationDbi_1.38.2
[100] memoise_1.1.0 IRanges_2.10.5 bindrcpp_0.2.2
[103] cluster_2.0.7-1
BTW:
I got the warning when
> library(scaffold)
Warning messages:
1: replacing previous import ‘flowCore::normalize’ by ‘igraph::normalize’ when loading ‘scaffold’
2: replacing previous import ‘flowCore::tree’ by ‘igraph::tree’ when loading ‘scaffold’
3: replacing previous import ‘flowCore::parent’ by ‘igraph::parent’ when loading ‘scaffold’
4: replacing previous import ‘plyr::rename’ by ‘reshape::rename’ when loading ‘scaffold’
5: replacing previous import ‘plyr::round_any’ by ‘reshape::round_any’ when loading ‘scaffold’
> scaffold.run()
Loading required package: shiny
Warning: package ‘shiny’ was built under R version 3.4.4
since zina commented here:
#8
I assumed that this should not be the cause of the issue.
Hello!
Everything has gone well with my first stab at scaffold so far, but I've run into the same issue three times now. All the data has been processed under the "run clustering" tab, but then I move to the "run scaffold analysis" step and select the first fcs file and the page goes dark and says it's processing, but it just stays like this for over an hour... anyone else experience this issue? Does this step usually take a long time? Thanks!
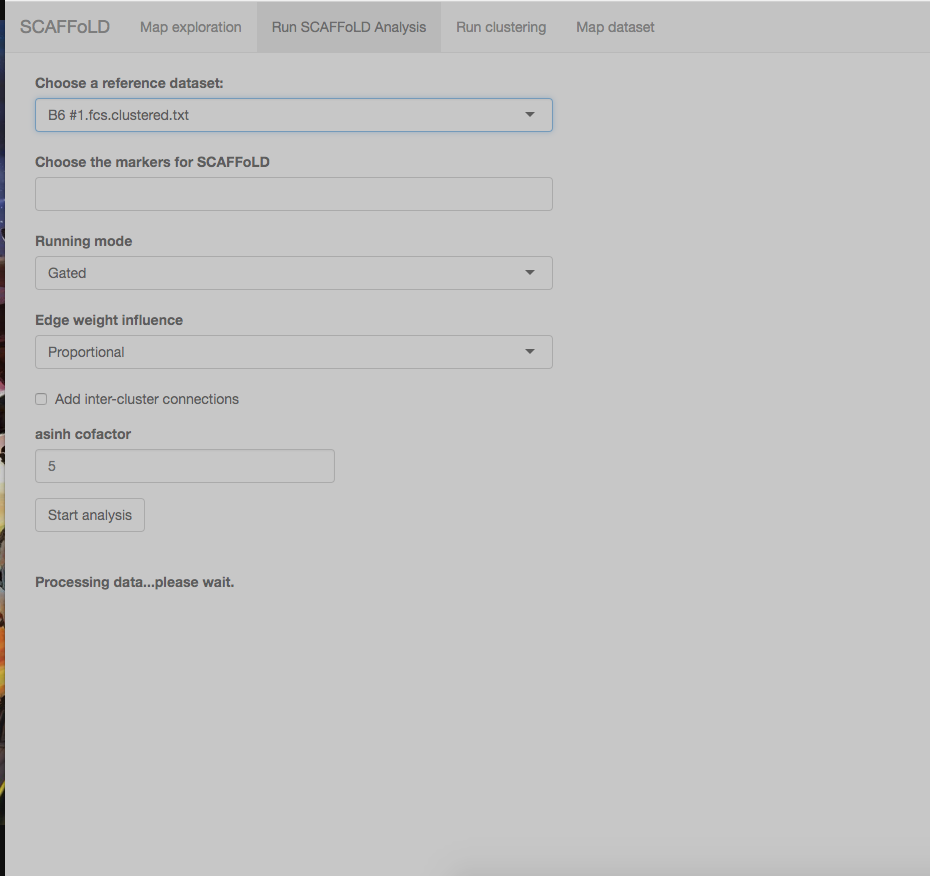
Hi there,
This is the first time I am using scaffold. I have got the .Rdata files and am trying to run the scaffold analysis as in the README. I keep getting an error "missing value where TRUE/FALSE needed". I have tried filling in different combinations of things in the drop down menu/tick boxes but still get this error. I have my gated fcs files in a subdirectory "gated"...
This is the print out I get in R studio -
tack trace (innermost first):
89: load_attractors_from_gated_data
88: scaffold:::run_analysis_gated
81: isolate
80: renderText [/Library/Frameworks/R.framework/Versions/3.3/Resources/library/scaffold/shinyGUI/server.R#352]
79: func
78: origRenderFunc
77: output$analysisui_empty
2: runApp
1: scaffold.run
Any suggestions where I am missing this TRUE/FALSE value?
Thanks very much.
Kirsten
PS. Sorry for cross posting - I initially put this in the wrong thread.
Hello
We are having issues using scaffold. After completing the clustering step, we get the usual message of "data processing is completed. However, neither the clustered.txt or the RData file are being created. We tried different datasets, different computers but no success yet. Any idea what could be wrong?
Hello,
I am currently working on SCAFFoLD (multifiles version) a bit and I ran into an error :
"_scaffold_layout_forceatlas2Cpp" not available for .Call() for package "scaffold"
I only have this error on 3.5+ R versions, so I assume this is a compatibility issue with it or the new versions of the packages used to run SCAFFoLD. I know you might not be working on this version anymore, but I share this bug in case someone else runs into it.
We are running SCAFFoLD for the first time.
We can successfully Run Clustering (files are generated) but when we try to Run SCAFFoLD Analysis we get an error message (Error: attempt to set an attribute on NULL). We have tried selecting different samples and marker combinations but same message.
Could you suggest what we need to change, please?
Thank you!
Hi,
I have got a set of FCS files that are already compensated and linearised. But the asinh transformation is mandatory. I think it would be possible to postulate that a zero coefficient means no transformation and modify the convert_fcs function accordingly. What's your point of view?
Samuel
Hi,
My team is having trouble running the SCAFFoLD package in R. We are using R Studio 3.4.4.
The GUI returns a successful completion message, but the warning message in R Studio is:
Warning in parallel::mclapply(files.list, mc.cores = num.cores, mc.preschedule = FALSE, :
21 function calls resulted in an error
Is this an issue with the SCAFFoLD package, the Bioconductor packages, or something else?
Thank you!
-Courtney
Hello,
I was trying to get SCAFFOLD to work again, but I'm still having issues. I have selected FCS files after entering the command "scaffold.run()", but when SCAFFOLD window opens on Chrome I can't select anything and thus can't run the analysis. Is anyone else having issues with the newest version of R? I was able to get a bit further into the analysis without running into problems on the previous version.
Thanks
Hello,
I've been asked to investigate operational aspects of scaffold. I'm missing a release of this.
Would you consider this stable software or could you point me to a stable commit/tag that provides reproducible versions of scaffold?
Thanks,
Martin
Dear Pier,
How can I export pdf's of the scaffold maps? Does it have to be via R?
Best,
Dunja
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.