greenelab / tybalt Goto Github PK
View Code? Open in Web Editor NEWTraining and evaluating a variational autoencoder for pan-cancer gene expression data
License: BSD 3-Clause "New" or "Revised" License
Training and evaluating a variational autoencoder for pan-cancer gene expression data
License: BSD 3-Clause "New" or "Revised" License
This has caused issues in the past (see #63) and was again brought up in greenelab/pancancer#59
I will need to revisit how features are named. This could be as simple as node_algorithm format. For example, ADAGE feature 17 would be 17_adage but could ease frustration and prevent breakage in the future.
Visualizing how each loss function contributes to the overall loss and how each is learned through each training epoch will be important. It will be important especially if warmup is implemented in #12
In #95 I removed outliers from visualizations to focus on areas of highest density. I defined outliers by > 3 z score for skew or kurtosis. The features appear to be identifiying similar biological processes across algorithms (e.g. patient sex). I need to decide if focusing on biological assignment for these features should be handled separately than other features.
this will inform any customized batching requirements to enforce balanced learning
I will need to establish a specific hypothesis and test latent space arithmetic in this context. This analysis may be an extension of the high grade serous ovarian cancer analyses presented in latent_space_comparison.ipynb
hi,
Which part of the code is to achieve T-SNE visualization of all tumor VAE coding features? I didn't find it in the project.
As @dhimmel brought up in greenelab/pancancer#56 some of the files used are large and should be compressed to save space and speed up access. Once the git LFS data are compressed and better organized, this issue will be resolved.
I'll use a couple issues to discuss capabilities/challenges/evaluation tactics
I need to perform a dimesionality reduction algorithm analysis of reconstruction costs. I will slowly increase the number of latent space features and observe how this relates to reconstruction cost for several algorithms.
This is much easier to do for PCA, ICA, and NMF. It will require a substantial refactoring of tybalt (see #74) and adage in order to facilitate these keras based models
This will be in anticipation of a large parameter sweep to be run on GPU cluster. I will need to determine supported versions
Code is getting unwieldy with the same functions and classes being defined in multiple places. There is also necessarily a lot of customization required to run each model. This issue will include information on how a refactoring should be performed.
Points to consider:
High weight gene selection performed naively by selecting the top n weight genes.
as @jaclyn-taroni pointed out in #95 - this method should be implemented in module tybalt.genes.high_weight_genes and be evaluated in upstream pathway analyses.
Hi,
I’m interested in the part that you have investigated the biological relevance of extracted latent features. However, I have not been bale to figure where you find the dimensions that are say most important in distinguishing patients' sex. It appears that this is feature 82. Is this feature 82 (out of 100) in the latent space of VAEs with one hidden layer, or two hidden layer? I’m guessing it’s the latter, which is why I was looking into tybalt_twohidden.ipynb. However, here only weights for all samples are extracted based on multiplying the weights of the two hidden layers. So the question I was hoping you can help me with is in the vae with two hidden layer, how the latent feature(s) that distinguishes patients's sex is identified as 82?
Hi Dr. Greg,
I tried to implement your code but was trapped when creating environment using environment.yml you provided. There are some conflicts for the dependency. I am not sure whether it is rasied by the reason the version might be too old. Will you update the dependencies?
Thanks
yurui
As discussed in Sonderby et al. 2016 a warm up period from deterministic to stochastic autoencoder model as training epochs proceed may enhance parameter usage.
This stackoverflow post may be useful to review.
I will use this issue to discuss which parameter combos to sweep over
Introduced in #103 - a simulation experiment is worth exploring. Currently, the simulation experiment samples the following signals (copy and pasted from #103):
As mentioned, some of the evals envisioned include:
Group 1: Yes Feature - Group 1: No Feature + Group 2: No Feature = Group 2: Yes Feature?)As suggested by @jaclyn-taroni, we will use this issue to track important citations and discuss rationale for the simulations and evals.
The file (encoded_adage_features.tsv) as stored on git is only three lines. tsne_tybalt_features.ipynb expects a true tsv file and throws a value error. I ran adage_training.ipynb on my machine and it made a working copy of the file.
Dear Dr. Greg,
I was trying to run your process_data.py script in nbconverted scripts. This resulted in an error, which I believe was because of the missing column 'hugoSymbol' in tsg_df and also because that indexing with list with missing labels is deprecated. Please find the exact output below. The documentation says:
Changed in version 1.0.0.
Using .loc or [] with a list with one or more missing labels will no longer reindex, in favor of .reindex.
Could I simply use .reindex() instead of .loc() here in this case? I would be very grateful for your help with this issue. Thank you.
Kind regards,
Yagmur
Passing list-likes to .loc or [] with any missing label will raise
KeyError in the future, you can use .reindex() as an alternative.
See the documentation here:
http://pandas.pydata.org/pandas-docs/stable/indexing.html#deprecate-loc-reindex-listlike
status_gain = copy_gain_df.loc[:, oncogenes_df['hugoSymbol']]
/mnt/lsf-nas-1/os-shared/anaconda3/envs/tybalt/lib/python3.5/site-packages/pandas/core/indexing.py:1367: FutureWarning:
Passing list-likes to .loc or [] with any missing label will raise
KeyError in the future, you can use .reindex() as an alternative.
See the documentation here:
http://pandas.pydata.org/pandas-docs/stable/indexing.html#deprecate-loc-reindex-listlike
return self._getitem_tuple(key)
tybalt/scripts/nbconverted/process_data.py:255: FutureWarning:
Passing list-likes to .loc or [] with any missing label will raise
KeyError in the future, you can use .reindex() as an alternative.
See the documentation here:
http://pandas.pydata.org/pandas-docs/stable/indexing.html#deprecate-loc-reindex-listlike
status_loss = copy_loss_df.loc[:, tsg_df['hugoSymbol']]
tybalt/scripts/nbconverted/process_data.py:270: FutureWarning:
Passing list-likes to .loc or [] with any missing label will raise
KeyError in the future, you can use .reindex() as an alternative.
See the documentation here:
http://pandas.pydata.org/pandas-docs/stable/indexing.html#deprecate-loc-reindex-listlike
copy_status = copy_status.loc[:, mutation_status.columns].fillna(0).astype(int)```
Hello,
I've essentially copied your tybalt_vae.ipynb file line for line in a new file to train on some of my own data, but after training on my dataset, it appears the training isn't occurring at all:
However, the code seems to function as intended otherwise (e..g no errors). I've noted that your training data is scaled [0,1], while mine is scaled to be centered on 0, with unit variance (e.g. not [0,1])--could this be the issue?
Distinct analyses are becoming confusing to follow. There are many parameter sweeps and simulation evaluations spanning compression models and latent space dimensionality.
The tybalt folder will become a standalone package, and the analyses that live in this repository must be easier to understand, especially as the project expands.
I'll use a couple issues to discuss capabilities/challenges/evaluation tactics
Initial ADAGE models allowed encoder and decoder weights to vary independently. This resulted in relatively poor performance in comparison to other models in simulated and real data reconstruction tasks.
Recent implementations (#123 , #126) have added an option to tie ADAGE weights together (decoder weights are transposed encoder weights). This improved performance. However, the optimal noise parameter observed in a parameter sweep for the pancanatlas data is noise = 0.

Based on previous observations, this should not be the case! We need to look into this in more detail. Currently, my thoughts are either:
tybalt_vae.ipynb currently trains a VAE on pancan data and outputs the latent layer along with compiled Keras models.
the notebook also does a few sanity checks to ensure the model is actually learning something. This includes ensuring there are no zeroed out features and a quick check on reconstruction fidelity.
A more thorough diagnostic notebook would help ensure robust training. I will use this issue to discuss what should be included in the future nb.
Setup Git LFS for future imports of large data files and models
Hi Greg,
Just a quick comment on something I spotted: in the manuscript you mentioned you've used median absolute deviation (MAD), but in the process_data.ipynb notebook you seem to use the pd.DataFrame.mad() method which, according to the docs, calculates the mean absolute deviation: https://pandas.pydata.org/pandas-docs/stable/generated/pandas.DataFrame.mad.html
Hope this helps (and sorry if I got that wrong!),
Enrico
add logging solution for std out and std err output for pmacs
This involves data simulation and interpolating between cancer-states. I need to define and test a hypothesis for this evaluation
this will be the workhorse of the repository - will need to take in command arguments and run through model training and preliminary evaluation.
By preliminary evaluation, I mean it will need to determine quickly which parameter combination is best, and output some sort of metric. I am thinking the metrics should be a combination of lowest loss and biological interpretability.
Hi Greg,
Thanks for all the work in putting this together. I'm trying to reproduce some of the results, and can't quite seem to get the separation that is shown in the paper.
At the simplest, I'm loading the existing results saved in encoded_rnaseq_onehidden_warmup_batchnorm.tsv, and then plotting:
plt.figure(figsize=(6, 6))
plt.scatter(encoded_rnaseq_df.iloc[:, 1], encoded_rnaseq_df.iloc[:, 2])
plt.xlabel('Latent Feature 1')
plt.ylabel('Latent Feature 2');(assuming line 4 above should be plt.ylabel) which produces:

Which seems to have less separation than the sanity check in the IPython notebook. Similarly when I plot feature 53 against 66:
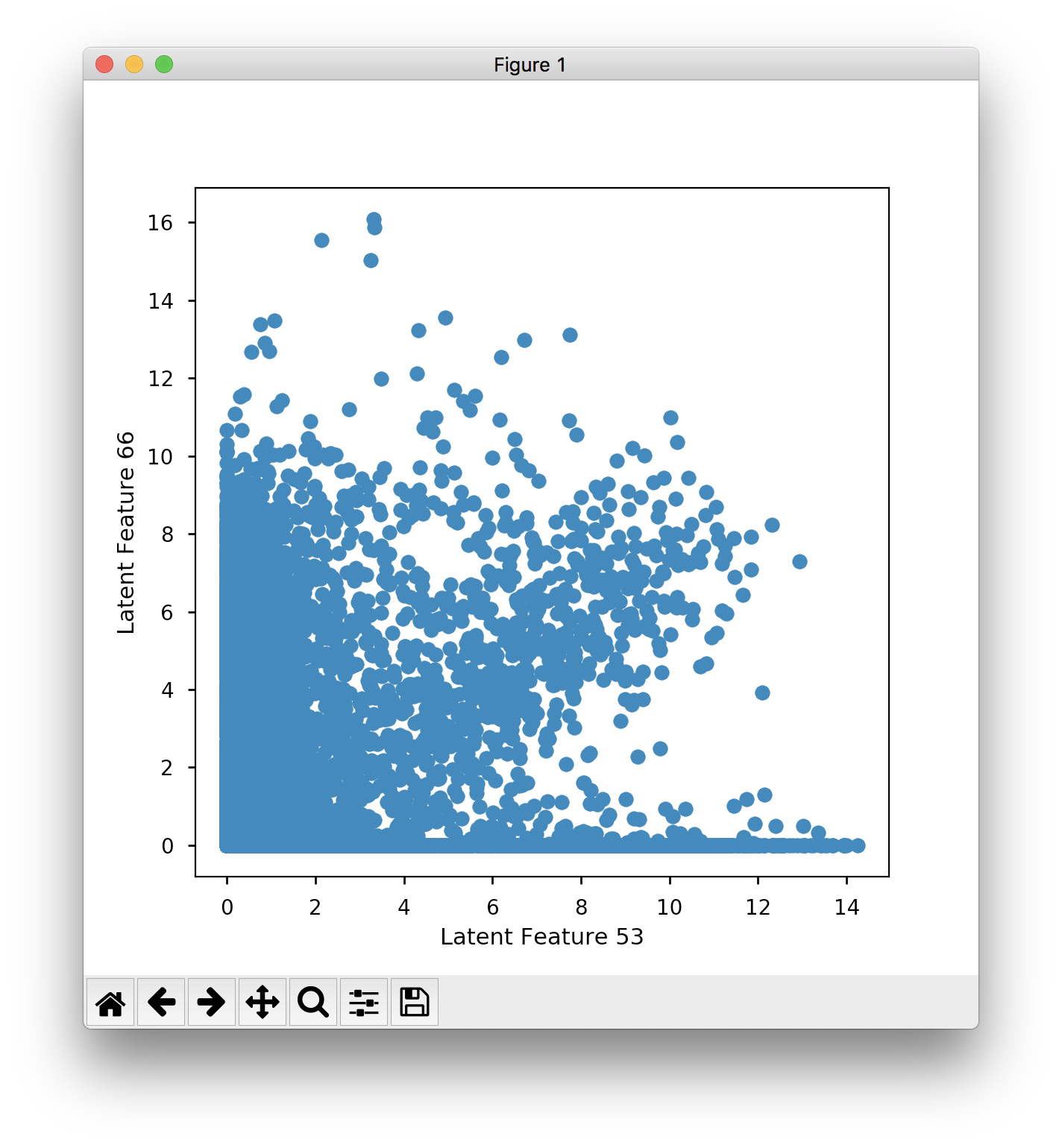
You can kind of see the cluster, but there is less separation than what is produced in Fig. 3B in the paper. When I train the model from scratch to produce the embeddings, I get similar separation to the saved results, and not the clear separation that you see in the papers.
Am I missing something?
the ADAGE gene weights (saved in cell 17 of adage_training.ipynb) are not saved in the same format as the Tybalt weights. This needs to be addressed.
I will need to determine a consistent file format to extract all high weight genes from each feature encoding.
This will help to facilitate some sort of pathway coverage analysis aiding in an alternative dimensionality reduction algorithm eval.
Hi Gregory,
I am very happy to see how you have detailed the steps for gene expression VAE based model. I am doing some analysis on gene expression prediction model to classify dementia. I started learning teh applications of VAE model/ machine learning models in omic prediction models. Also I am new to Tensorflow/Keras.
I successfully implemented your code using your gene expression data after disabling the eager execution to make your program work in Tensorflow 2. But when we use our gene expression data (which is log 2 cpm normalized data), I am getting all the reconstructed values as 1.0 so my gene_mean and gene_summary remains same. I evaluated your data distribution ( it looks gene expression data in the range of 0-1).
Could you let me know if you can think of any issue with my input data shape.
Input:
Dimension: (3045, 10956)
data:
| XXbac-BPG248L24.12 | TTN | RP11-290D2.6 | JSRP1 | RP11-115D19.1 | HCG4P5 | AC114271.2 | RP3-394A18.1 | ABALON | KB-1208A12.3 | ... | LNPK | NBPF15 | ATP8B4 | AC005522.7 | CHID1 | ARFRP1 | NAPB | CTB-133G6.2 | SPATA24 | POU2F2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12.988314 | 12.83923 | 12.576047 | 12.136305 | 11.978625 | 12.494600 | 12.583452 | 12.415211 | 10.807796 | 11.77091 | ... | 3.774915 | 3.350120 | 3.774915 | 2.310416 | 1.684627 | 2.745442 | 3.350120 | 2.745442 | 2.310416 | 3.350120 |
| 12.641275 | 12.56024 | 12.252506 | 12.576744 | 12.883745 | 12.327777 | 11.751295 | 11.776363 | 12.345054 | 11.64166 | ... | 2.240370 | 2.953526 | 3.618031 |
input_rnaseq_reconstruct.head(2):
| XXbac-BPG248L24.12 | TTN | RP11-290D2.6 | JSRP1 | RP11-115D19.1 | HCG4P5 | AC114271.2 | RP3-394A18.1 | ABALON | KB-1208A12.3 | ... | LNPK | NBPF15 | ATP8B4 | AC005522.7 | CHID1 | ARFRP1 | NAPB | CTB-133G6.2 | SPATA24 | POU2F2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | ... | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | ... | 1.0 | 1.0 | 1.0 | 1.0 |
gene_summary:
| gene mean | gene abs(sum) |
|---|---|
| 11.867037 | 11.867037 |
| 11.774692 | 11.774692 |
| 11.622958 | 11.622958 |
| 11.489614 | 11.489614 |
| 11.474622 | 11.474622 |
Sorry for all the Issues, but this should be the last one for a while:
Is there an efficient manner to sample the latent space? I'm currently just randomly sampling the range of min/max for each feature, and having the decoder give the proper representation for that latent feature set, but not sure if thats the best route.
EDIT: To clarify, I have a classifier that can distinguish two populations based on rnaseq data. I would like to iteratively go through each gene in my model and see what happens when it is changed/lowered to a specific range. To do this, I'd like to sample the target distribution (for that given gene; likely just a 1sigma interval of 'normal' expression) and then run my classifier on the backend. Given the non-linear dependency of all the other genes on the target gene, the idea is to just randomly sample the latent space(and the learned distributions for each gene), and just select those vectors whose values meet my 1sigma criterion (on a per-gene basis, and hope that the representations of the other genes "respond" to the change in expression on my target gene).
Hey, great work on this project! I'm try to re-purpose this for something a little different. After training the VAE, I would like to focus on particular genes, and sample around a specific range, and see how that affects other gene distributions. Is that at all possible?
A good indicator of "how much biology did the model learn" is based on a pathway coverage test. I need to see how this was done in the eADAGE paper and then perform the pathway coverage on Pan-Cancer data for dimensionality reduction algorithms: PCA, ICA, NMF, ADAGE, and Tybalt.
Is there a better pathway database for cancer? Perhaps something like KEGG cancer?
I will want to save the tybalt weights as a separate file on the Master branch
Hi @gwaygenomics @cgreene
I would like to map samples (index values) in pancan_scaled_rnaseq.tsv.gz to the metadata tcga-clinical_data.tsv.
Currently the index values for the rnaseq are not unique and I am unable to match them to the metadata.
Could you please advise on how, for example, I could subset the pancancer data to just a single cancer subtype (tying it to the metadata).
In extract_tybalt_weights.ipynb notebook, you defined a function output_high_weight_genes.
At a certain point you have these 2 lines:
hw_pos_df = pd.DataFrame(encoding_df[encoding_df > encoding_df.std() * thresh])
hw_neg_df = pd.DataFrame(encoding_df[encoding_df < -encoding_df.std() * thresh])
It seems like you are trying to get the most extreme values away from the mean, but you don't really have the mean here. Shouldn't it be something like this?
hw_pos_df = pd.DataFrame(encoding_df[encoding_df > encoding_df.mean() + encoding_df.std() * thresh])
hw_neg_df = pd.DataFrame(encoding_df[encoding_df < encoding_df.mean() - encoding_df.std() * thresh])
Dear Dr. Greg,
Firstly, thank you very much for this well-documented research. This is not an issue but a feature that I need for my thesis. I would like to modify this model so that it can handle missing values in the test and training data. Because the data set I want to use in the future contains missing values in every sample. Imputing missing values before the training e.g. with zeros is not an option as this would introduce bias and the value zero has a meaning in our data set. Therefore I started with replacing the reconstruction error function with my custom binary cross-entropy function that can handle missing values. I tested this function in another notebook and it seemed to work. However, I observe NaN loss values in the training, also even after only using the reconstruction error. If you have any tips for me regarding how to handle missing values in the model, I would be very grateful for your help.
Kind regards,
Yagmur Onay
Error using Keras version 2.1.3 and tensorflow version 1.5.0
The notebook I uploaded doesn't have the full training results... I need to rerun. Also should rerun with magic time
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.