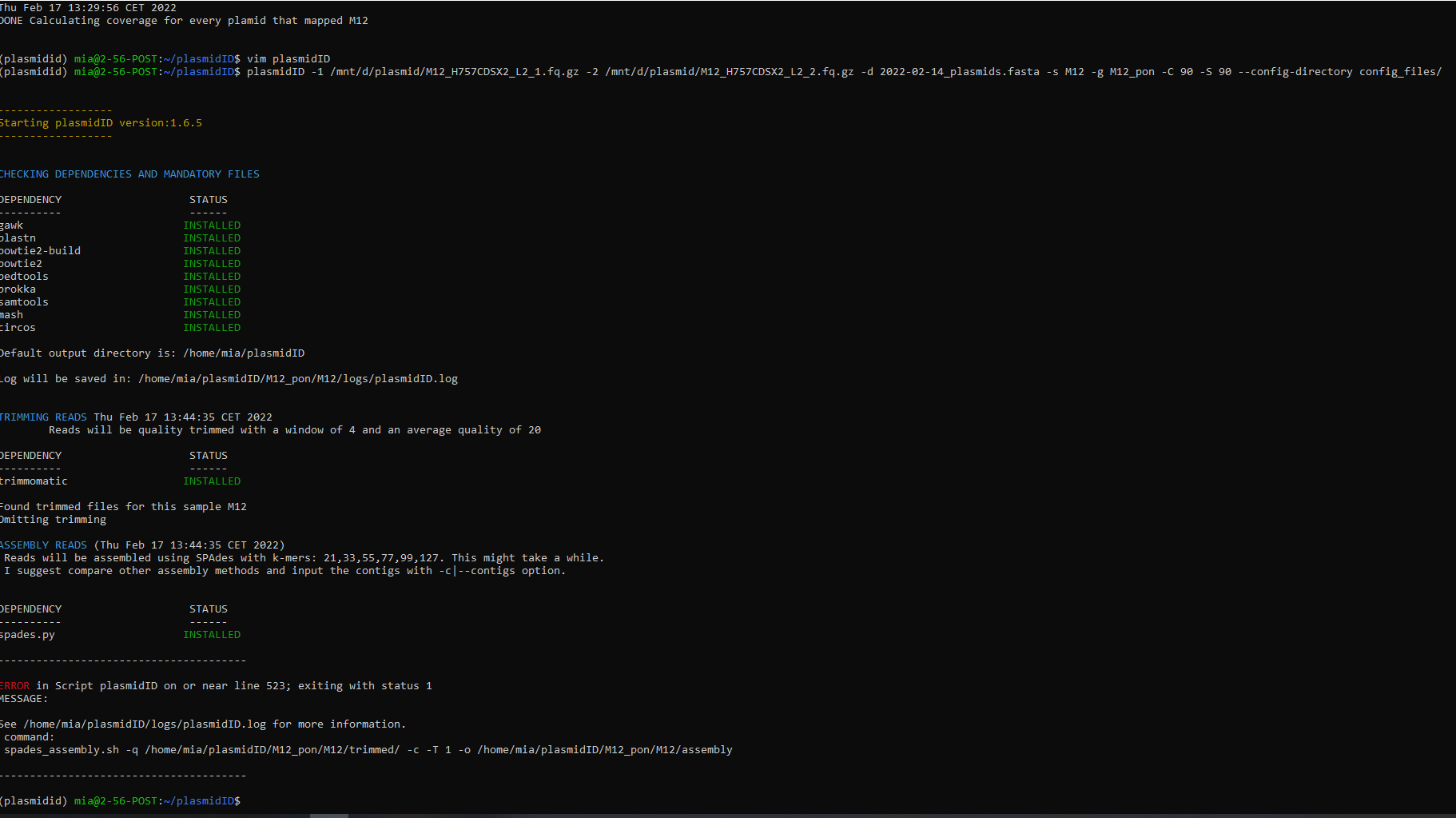
Hi,
How are you? I hope this email finds you well!
I've been trying to run this tool to predict plasmids in e. coli, but I've seen to have run into a problem. It seems to be some kind of problem with the sorted.bam file (according to detailed in the log file). I installed plasmidID using conda, as detailed in the instructions.
Any help will be greatly appreciated! Thanks a lot for your help!
I detailed below the output from the tool and the log
- OUTPUT FROM THE TOOL
(plasmid_id) [jpaganini@n0061 reads]$ plasmidID -1 reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq -2 reads/GCA_011404755.1_ASM1140475v1_genomic_R2.fq -d plasmidid_db/2020-08-19_plasmids.fasta -s GCA_011404755.1_ASM1140475v1_genomic -o 20200830_test_output
Starting plasmidID version:1.6.3
CHECKING DEPENDENCIES AND MANDATORY FILES
DEPENDENCY STATUS
blastn INSTALLED
bowtie2-build INSTALLED
bowtie2 INSTALLED
bedtools INSTALLED
prokka INSTALLED
samtools INSTALLED
mash INSTALLED
circos INSTALLED
GCA_011404755.1_ASM1140475v1_genomic_R1.fq not supplied, please, introduce a valid file
GCA_011404755.1_ASM1140475v1_genomic_R2.fq not supplied, please, introduce a valid file
2020-08-19_plasmids.fasta not supplied, please, introduce a valid file
ERROR: 3 missing files, aborting execution
(plasmid_id) [jpaganini@n0061 reads]$ cd ..
(plasmid_id) [jpaganini@n0061 testing_plasmidid]$ plasmidID -1 reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq -2 reads/GCA_011404755.1_ASM1140475v1_genomic_R2.fq -d plasmidid_db/2020-08-19_plasmids.fasta -s GCA_011404755.1_ASM1140475v1_genomic -o 20200830_test_output
Starting plasmidID version:1.6.3
CHECKING DEPENDENCIES AND MANDATORY FILES
DEPENDENCY STATUS
blastn INSTALLED
bowtie2-build INSTALLED
bowtie2 INSTALLED
bedtools INSTALLED
prokka INSTALLED
samtools INSTALLED
mash INSTALLED
circos INSTALLED
Output directory is: 20200830_test_output
Log will be saved in: 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/logs/plasmidID.log
TRIMMING READS Sun Aug 30 13:46:12 CEST 2020
Reads will be quality trimmed with a window of 4 and an average quality of 20
DEPENDENCY STATUS
trimmomatic INSTALLED
ASSEMBLY READS (Sun Aug 30 13:52:39 CEST 2020)
Reads will be assembled using SPAdes with k-mers: 21,33,55,77,99,127. This might take a while.
I suggest compare other assembly methods and input the contigs with -c|--contigs option.
DEPENDENCY STATUS
spades.py INSTALLED
#Pipeline summary#
Reads R1 GCA_011404755.1_ASM1140475v1_genomic_R1.fq
Reads R2 GCA_011404755.1_ASM1140475v1_genomic_R2.fq
Will be mapped with ddbb 2020-08-19_plasmids.fasta
Entries covered more than 80 %
Will be clustered by 0.5 % identity
And used to reconstruct contigs in scaffolds.fasta
STARTING KMER FILTERING, CLUSTERING and MAPPING
SCREENING READS WITH KMERS (Sun Aug 30 14:40:09 CEST 2020)
Reads will be screened against database supplied for further filtering and mapping,
this will reduce the input sequences to map against GCA_011404755.1_ASM1140475v1_genomic
CLUSTERING SEQUENCES BY KMER DISTANCE (Sun Aug 30 14:45:57 CEST 2020)
Sequences obtained after screen will be clustered to reduce redundancy,
one representative, the largest, will be considered for further analysis GCA_011404755.1_ASM1140475v1_genomic
MAPPING READS (Sun Aug 30 14:46:09 CEST 2020)
Reads will be mapped against database supplied for further coverage calculation,
this will determine the most likely plasmids in the sample GCA_011404755.1_ASM1140475v1_genomic
FILTERING DATABASE BY COVERAGE (Sun Aug 30 14:52:56 CEST 2020)
Coverage will be calculated and the entries covered more than 80%
will pass to further analysis
ERROR in Script plasmidID on or near line 642; exiting with status 1
MESSAGE:
See 20200830_test_output/logs/plasmidID.log for more information.
command:
get_coverage.sh -i 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/mapping/GCA_011404755.1_ASM1140475v1_genomic".sorted.bam" -d plasmidid_db/2020-08-19_plasmids.fasta
################################################################################################
- LOG FILE:
LOG FILE PLASMIDID
Sun Aug 30 13:46:12 CEST 2020
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/quality_trim.sh
DEPENDENCY STATUS
trimmomatic �[0;32mINSTALLED�[0m
Output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed
Sun Aug 30 13:50:38 CEST 2020
Quality trimming:
R1 = reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq
R2 = reads/GCA_011404755.1_ASM1140475v1_genomic_R2.fq
TrimmomaticPE: Started with arguments:
-threads 1 reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq reads/GCA_011404755.1_ASM1140475v1_genomic_R2.fq 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_unpaired.fastq.gz 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_unpaired.fastq.gz ILLUMINACLIP:/hpc/dla_mm/jpaganini/data/miniconda3/pkgs/trimmomatic-0.39-1/share/trimmomatic-0.39-1/adapters/TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:40
Using PrefixPair: 'TACACTCTTTCCCTACACGACGCTCTTCCGATCT' and 'GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT'
ILLUMINACLIP: Using 1 prefix pairs, 0 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Quality encoding detected as phred33
Input Read Pairs: 1000000 Both Surviving: 999995 (100.00%) Forward Only Surviving: 4 (0.00%) Reverse Only Surviving: 1 (0.00%) Dropped: 0 (0.00%)
TrimmomaticPE: Completed successfully
Sun Aug 30 13:52:39 CEST 2020
DONE quality trimming, file can be fount at:
20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz
20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_unpaired.fastq.gz
20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_unpaired.fastq.gz
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/spades_assembly.sh
DEPENDENCY STATUS
spades.py �[0;32mINSTALLED�[0m
Reads directory for quick mode is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/
Output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly
Entering QUICK MODE
Sun Aug 30 13:52:39 CEST 2020
Assembly:
R1 paired file = 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz
R2 paired file = 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
Command line: /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/spades.py --careful -t 1 -k 21,33,55,77,99,127 --pe1-1 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz --pe1-2 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz -o /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly
System information:
SPAdes version: 3.13.0
Python version: 3.6.6
OS: Linux-3.10.0-1062.4.1.el7.x86_64-x86_64-with-centos-7.7.1908-Core
Output dir: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly
Mode: read error correction and assembling
Debug mode is turned OFF
Dataset parameters:
Multi-cell mode (you should set '--sc' flag if input data was obtained with MDA (single-cell) technology or --meta flag if processing metagenomic dataset)
Reads:
Library number: 1, library type: paired-end
orientation: fr
left reads: ['/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz']
right reads: ['/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz']
interlaced reads: not specified
single reads: not specified
merged reads: not specified
Read error correction parameters:
Iterations: 1
PHRED offset will be auto-detected
Corrected reads will be compressed
Assembly parameters:
k: [21, 33, 55, 77, 99, 127]
Repeat resolution is enabled
Mismatch careful mode is turned ON
MismatchCorrector will be used
Coverage cutoff is turned OFF
Other parameters:
Dir for temp files: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/tmp
Threads: 1
Memory limit (in Gb): 250
======= SPAdes pipeline started. Log can be found here: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/spades.log
===== Read error correction started.
== Running read error correction tool: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-hammer /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/corrected/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 75) Starting BayesHammer, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 76) Loading config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/corrected/configs/config.info
0:00:00.001 4M / 4M INFO General (main.cpp : 78) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.001 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.001 4M / 4M INFO General (main.cpp : 86) Trying to determine PHRED offset
0:00:00.002 4M / 4M INFO General (main.cpp : 92) Determined value is 33
0:00:00.003 4M / 4M INFO General (hammer_tools.cpp : 36) Hamming graph threshold tau=1, k=21, subkmer positions = [ 0 10 ]
0:00:00.003 4M / 4M INFO General (main.cpp : 113) Size of aux. kmer data 24 bytes
=== ITERATION 0 begins ===
0:00:00.007 4M / 4M INFO K-mer Index Building (kmer_index_builder.hpp : 301) Building kmer index
0:00:00.007 4M / 4M INFO General (kmer_index_builder.hpp : 117) Splitting kmer instances into 16 files using 1 threads. This might take a while.
0:00:00.009 4M / 4M INFO General (file_limit.hpp : 32) Open file limit set to 4096
0:00:00.009 4M / 4M INFO General (kmer_splitters.hpp : 89) Memory available for splitting buffers: 13.332 Gb
0:00:00.009 4M / 4M INFO General (kmer_splitters.hpp : 97) Using cell size of 4194304
0:00:00.009 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 97) Processing /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz
0:00:14.156 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 256726 reads
0:00:28.085 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 513421 reads
0:00:41.771 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 770008 reads
0:00:54.436 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 999995 reads
0:00:54.436 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 97) Processing /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
0:01:08.126 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 1256743 reads
0:01:22.308 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 107) Processed 1513407 reads
0:01:48.806 580M / 616M INFO K-mer Splitting (kmer_data.cpp : 112) Total 1999990 reads processed
0:01:48.878 4M / 616M INFO General (kmer_index_builder.hpp : 120) Starting k-mer counting.
0:01:57.428 4M / 616M INFO General (kmer_index_builder.hpp : 127) K-mer counting done. There are 44205208 kmers in total.
0:01:57.428 4M / 616M INFO General (kmer_index_builder.hpp : 133) Merging temporary buckets.
0:02:00.710 4M / 616M INFO K-mer Index Building (kmer_index_builder.hpp : 314) Building perfect hash indices
0:02:08.439 24M / 616M INFO General (kmer_index_builder.hpp : 150) Merging final buckets.
0:02:11.668 24M / 616M INFO K-mer Index Building (kmer_index_builder.hpp : 336) Index built. Total 20506240 bytes occupied (3.7111 bits per kmer).
0:02:11.671 24M / 616M INFO K-mer Counting (kmer_data.cpp : 356) Arranging kmers in hash map order
0:02:17.015 704M / 704M INFO General (main.cpp : 148) Clustering Hamming graph.
0:08:43.944 704M / 704M INFO General (main.cpp : 155) Extracting clusters
0:09:22.040 704M / 1G INFO General (main.cpp : 167) Clustering done. Total clusters: 11067046
0:09:22.073 364M / 1G INFO K-mer Counting (kmer_data.cpp : 376) Collecting K-mer information, this takes a while.
0:09:22.694 1G / 1G INFO K-mer Counting (kmer_data.cpp : 382) Processing /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz
0:11:52.857 1G / 1G INFO K-mer Counting (kmer_data.cpp : 382) Processing /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
0:14:21.757 1G / 1G INFO K-mer Counting (kmer_data.cpp : 389) Collection done, postprocessing.
0:14:21.932 1G / 1G INFO K-mer Counting (kmer_data.cpp : 403) There are 44205208 kmers in total. Among them 34158964 (77.2736%) are singletons.
0:14:21.932 1G / 1G INFO General (main.cpp : 173) Subclustering Hamming graph
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 649) Subclustering done. Total 22 non-read kmers were generated.
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 650) Subclustering statistics:
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 651) Total singleton hamming clusters: 2471282. Among them 828412 (33.5215%) are good
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 652) Total singleton subclusters: 1719. Among them 1681 (97.7894%) are good
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 653) Total non-singleton subcluster centers: 8673370. Among them 8641493 (99.6325%) are good
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 654) Average size of non-trivial subcluster: 4.81173 kmers
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 655) Average number of sub-clusters per non-singleton cluster: 1.00923
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 656) Total solid k-mers: 9471586
0:15:21.345 1G / 1G INFO Hamming Subclustering (kmer_cluster.cpp : 657) Substitution probabilities: 4,4
0:15:21.361 1G / 1G INFO General (main.cpp : 178) Finished clustering.
0:15:21.361 1G / 1G INFO General (main.cpp : 197) Starting solid k-mers expansion in 1 threads.
0:16:56.911 1G / 1G INFO General (main.cpp : 218) Solid k-mers iteration 0 produced 471136 new k-mers.
0:18:32.504 1G / 1G INFO General (main.cpp : 218) Solid k-mers iteration 1 produced 6399 new k-mers.
0:20:07.929 1G / 1G INFO General (main.cpp : 218) Solid k-mers iteration 2 produced 92 new k-mers.
0:21:43.504 1G / 1G INFO General (main.cpp : 218) Solid k-mers iteration 3 produced 0 new k-mers.
0:21:43.504 1G / 1G INFO General (main.cpp : 222) Solid k-mers finalized
0:21:43.504 1G / 1G INFO General (hammer_tools.cpp : 220) Starting read correction in 1 threads.
0:21:43.504 1G / 1G INFO General (hammer_tools.cpp : 233) Correcting pair of reads: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz and /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
0:21:44.282 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 0 of 100000 reads.
0:21:55.033 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 0
0:21:55.177 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 0
0:21:55.885 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 1 of 100000 reads.
0:22:06.635 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 1
0:22:06.781 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 1
0:22:07.481 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 2 of 100000 reads.
0:22:18.261 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 2
0:22:18.408 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 2
0:22:19.108 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 3 of 100000 reads.
0:22:29.872 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 3
0:22:30.018 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 3
0:22:30.721 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 4 of 100000 reads.
0:22:41.559 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 4
0:22:41.705 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 4
0:22:42.406 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 5 of 100000 reads.
0:22:53.146 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 5
0:22:53.290 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 5
0:22:53.994 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 6 of 100000 reads.
0:23:04.727 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 6
0:23:04.877 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 6
0:23:05.578 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 7 of 100000 reads.
0:23:16.342 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 7
0:23:16.491 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 7
0:23:17.192 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 8 of 100000 reads.
0:23:27.956 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 8
0:23:28.101 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 8
0:23:28.802 1G / 1G INFO General (hammer_tools.cpp : 168) Prepared batch 9 of 99995 reads.
0:23:39.562 1G / 1G INFO General (hammer_tools.cpp : 175) Processed batch 9
0:23:39.707 1G / 1G INFO General (hammer_tools.cpp : 185) Written batch 9
0:23:40.381 1G / 1G INFO General (hammer_tools.cpp : 274) Correction done. Changed 986783 bases in 805818 reads.
0:23:40.381 1G / 1G INFO General (hammer_tools.cpp : 275) Failed to correct 96864 bases out of 301903719.
0:23:40.485 4M / 1G INFO General (main.cpp : 255) Saving corrected dataset description to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/corrected/corrected.yaml
0:23:40.490 4M / 1G INFO General (main.cpp : 262) All done. Exiting.
== Compressing corrected reads (with pigz)
== Dataset description file was created: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/corrected/corrected.yaml
===== Read error correction finished.
===== Assembling started.
== Running assembler: K21
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K21/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K21/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/dataset.info) with K=21
0:00:00.000 4M / 4M INFO General (main.cpp : 90) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO General (launch.hpp : 51) SPAdes started
0:00:00.000 4M / 4M INFO General (launch.hpp : 58) Starting from stage: construction
0:00:00.000 4M / 4M INFO General (launch.hpp : 65) Two-step RR enabled: 0
0:00:00.001 4M / 4M INFO StageManager (stage.cpp : 132) STAGE == de Bruijn graph construction
0:00:00.004 4M / 4M INFO General (read_converter.hpp : 77) Converting reads to binary format for library #0 (takes a while)
0:00:00.004 4M / 4M INFO General (read_converter.hpp : 78) Converting paired reads
0:00:00.372 80M / 132M INFO General (binary_converter.hpp : 93) 16384 reads processed
0:00:00.555 96M / 132M INFO General (binary_converter.hpp : 93) 32768 reads processed
0:00:00.923 124M / 132M INFO General (binary_converter.hpp : 93) 65536 reads processed
0:00:01.661 180M / 180M INFO General (binary_converter.hpp : 93) 131072 reads processed
0:00:03.143 292M / 292M INFO General (binary_converter.hpp : 93) 262144 reads processed
0:00:06.101 520M / 520M INFO General (binary_converter.hpp : 93) 524288 reads processed
0:00:15.137 124M / 876M INFO General (binary_converter.hpp : 117) 999995 reads written
0:00:15.197 4M / 876M INFO General (read_converter.hpp : 87) Converting single reads
0:00:15.505 132M / 876M INFO General (binary_converter.hpp : 117) 0 reads written
0:00:15.518 4M / 876M INFO General (read_converter.hpp : 95) Converting merged reads
0:00:15.825 132M / 876M INFO General (binary_converter.hpp : 117) 0 reads written
0:00:16.694 4M / 876M INFO General (construction.cpp : 111) Max read length 151
0:00:16.695 4M / 876M INFO General (construction.cpp : 117) Average read length 150.933
0:00:16.695 4M / 876M INFO General (stage.cpp : 101) PROCEDURE == k+1-mer counting
0:00:16.697 4M / 876M INFO General (kmer_index_builder.hpp : 117) Splitting kmer instances into 1 files using 1 threads. This might take a while.
0:00:16.699 4M / 876M INFO General (file_limit.hpp : 32) Open file limit set to 4096
0:00:16.699 4M / 876M INFO General (kmer_splitters.hpp : 89) Memory available for splitting buffers: 13.332 Gb
0:00:16.699 4M / 876M INFO General (kmer_splitters.hpp : 97) Using cell size of 67108864
0:00:29.697 568M / 1G INFO General (kmer_splitters.hpp : 289) Processed 1032972 reads
0:00:42.515 568M / 1G INFO General (kmer_splitters.hpp : 289) Processed 2065962 reads
0:00:55.532 568M / 1G INFO General (kmer_splitters.hpp : 289) Processed 3098942 reads
0:01:07.105 568M / 1G INFO General (kmer_splitters.hpp : 289) Processed 3999980 reads
0:01:07.105 568M / 1G INFO General (kmer_splitters.hpp : 295) Adding contigs from previous K
0:01:07.148 4M / 1G INFO General (kmer_splitters.hpp : 308) Used 3999980 reads
0:01:07.148 4M / 1G INFO General (kmer_index_builder.hpp : 120) Starting k-mer counting.
0:01:08.357 4M / 1G INFO General (kmer_index_builder.hpp : 127) K-mer counting done. There are 6538497 kmers in total.
0:01:08.358 4M / 1G INFO General (kmer_index_builder.hpp : 133) Merging temporary buckets.
0:01:08.840 4M / 1G INFO General (stage.cpp : 101) PROCEDURE == Extension index construction
0:01:08.842 4M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 301) Building kmer index
0:01:08.842 4M / 1G INFO General (kmer_index_builder.hpp : 117) Splitting kmer instances into 16 files using 1 threads. This might take a while.
0:01:08.844 4M / 1G INFO General (file_limit.hpp : 32) Open file limit set to 4096
0:01:08.844 4M / 1G INFO General (kmer_splitters.hpp : 89) Memory available for splitting buffers: 13.332 Gb
0:01:08.844 4M / 1G INFO General (kmer_splitters.hpp : 97) Using cell size of 4194304
0:01:11.889 580M / 1G INFO General (kmer_splitters.hpp : 380) Processed 6538497 kmers
0:01:11.889 580M / 1G INFO General (kmer_splitters.hpp : 385) Used 6538497 kmers.
0:01:11.904 4M / 1G INFO General (kmer_index_builder.hpp : 120) Starting k-mer counting.
0:01:12.737 4M / 1G INFO General (kmer_index_builder.hpp : 127) K-mer counting done. There are 6494188 kmers in total.
0:01:12.737 4M / 1G INFO General (kmer_index_builder.hpp : 133) Merging temporary buckets.
0:01:13.314 4M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 314) Building perfect hash indices
0:01:14.434 4M / 1G INFO General (kmer_index_builder.hpp : 150) Merging final buckets.
0:01:14.922 4M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 336) Index built. Total 3019896 bytes occupied (3.72012 bits per kmer).
0:01:14.927 12M / 1G INFO DeBruijnExtensionIndexBu (kmer_extension_index_build: 99) Building k-mer extensions from k+1-mers
0:01:16.923 12M / 1G INFO DeBruijnExtensionIndexBu (kmer_extension_index_build: 103) Building k-mer extensions from k+1-mers finished.
0:01:16.925 12M / 1G INFO General (stage.cpp : 101) PROCEDURE == Early tip clipping
0:01:16.925 12M / 1G INFO General (construction.cpp : 253) Early tip clipper length bound set as (RL - K)
0:01:16.925 12M / 1G INFO Early tip clipping (early_simplification.hpp : 181) Early tip clipping
0:01:21.762 16M / 1G INFO Early tip clipping (early_simplification.hpp : 184) 997269 22-mers were removed by early tip clipper
0:01:21.762 16M / 1G INFO General (stage.cpp : 101) PROCEDURE == Condensing graph
0:01:21.765 16M / 1G INFO UnbranchingPathExtractor (debruijn_graph_constructor: 355) Extracting unbranching paths
0:01:24.640 24M / 1G INFO UnbranchingPathExtractor (debruijn_graph_constructor: 374) Extracting unbranching paths finished. 131049 sequences extracted
0:01:26.081 24M / 1G INFO UnbranchingPathExtractor (debruijn_graph_constructor: 310) Collecting perfect loops
0:01:26.769 24M / 1G INFO UnbranchingPathExtractor (debruijn_graph_constructor: 343) Collecting perfect loops finished. 0 loops collected
0:01:26.940 52M / 1G INFO General (stage.cpp : 101) PROCEDURE == Filling coverage indices (PHM)
0:01:26.940 52M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 301) Building kmer index
0:01:26.940 52M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 314) Building perfect hash indices
0:01:28.152 56M / 1G INFO K-mer Index Building (kmer_index_builder.hpp : 336) Index built. Total 3032416 bytes occupied (3.71023 bits per kmer).
0:01:28.165 84M / 1G INFO General (construction.cpp : 388) Collecting k-mer coverage information from reads, this takes a while.
0:02:09.252 80M / 1G INFO General (construction.cpp : 508) Filling coverage and flanking coverage from PHM
0:02:11.043 80M / 1G INFO General (construction.cpp : 464) Processed 262082 edges
0:02:11.086 40M / 1G INFO StageManager (stage.cpp : 132) STAGE == EC Threshold Finding
0:02:11.087 40M / 1G INFO General (kmer_coverage_model.cpp : 181) Kmer coverage valley at: 14
0:02:11.087 40M / 1G INFO General (kmer_coverage_model.cpp : 201) K-mer histogram maximum: 55
0:02:11.087 40M / 1G INFO General (kmer_coverage_model.cpp : 237) Estimated median coverage: 56. Coverage mad: 8.8956
0:02:11.087 40M / 1G INFO General (kmer_coverage_model.cpp : 259) Fitting coverage model
0:02:11.161 40M / 1G INFO General (kmer_coverage_model.cpp : 295) ... iteration 2
0:02:11.315 40M / 1G INFO General (kmer_coverage_model.cpp : 295) ... iteration 4
0:02:11.893 40M / 1G INFO General (kmer_coverage_model.cpp : 295) ... iteration 8
0:02:12.657 40M / 1G INFO General (kmer_coverage_model.cpp : 309) Fitted mean coverage: 56.5557. Fitted coverage std. dev: 7.58561
0:02:12.660 40M / 1G INFO General (kmer_coverage_model.cpp : 334) Probability of erroneous kmer at valley: 0.99998
0:02:12.660 40M / 1G INFO General (kmer_coverage_model.cpp : 358) Preliminary threshold calculated as: 35
0:02:12.660 40M / 1G INFO General (kmer_coverage_model.cpp : 362) Threshold adjusted to: 35
0:02:12.660 40M / 1G INFO General (kmer_coverage_model.cpp : 375) Estimated genome size (ignoring repeats): 4441797
0:02:12.660 40M / 1G INFO General (genomic_info_filler.cpp : 112) Mean coverage was calculated as 56.5557
0:02:12.660 40M / 1G INFO General (genomic_info_filler.cpp : 127) EC coverage threshold value was calculated as 35
0:02:12.660 40M / 1G INFO General (genomic_info_filler.cpp : 128) Trusted kmer low bound: 0
0:02:12.660 40M / 1G INFO StageManager (stage.cpp : 132) STAGE == Raw Simplification
0:02:12.660 40M / 1G INFO General (simplification.cpp : 128) PROCEDURE == InitialCleaning
0:02:12.660 40M / 1G INFO General (graph_simplification.hpp : 662) Flanking coverage based disconnection disabled
0:02:12.660 40M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Self conjugate edge remover
0:02:12.696 40M / 1G INFO Simplification (parallel_processing.hpp : 167) Self conjugate edge remover triggered 0 times
0:02:12.696 40M / 1G INFO StageManager (stage.cpp : 132) STAGE == Simplification
0:02:12.696 40M / 1G INFO General (simplification.cpp : 357) Graph simplification started
0:02:12.696 40M / 1G INFO General (graph_simplification.hpp : 634) Creating parallel br instance
0:02:12.696 40M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 1
0:02:12.696 40M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:12.741 40M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 174 times
0:02:12.741 40M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.111 40M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 41002 times
0:02:14.111 40M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.179 32M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 2083 times
0:02:14.179 32M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 2
0:02:14.179 32M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.183 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 17 times
0:02:14.183 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 76 times
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 20 times
0:02:14.196 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 3
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.196 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 5 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 4
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 5
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 6
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 7
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 8
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 9
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 10
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 1 times
0:02:14.199 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 11
0:02:14.199 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.200 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 1 times
0:02:14.200 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.211 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 7 times
0:02:14.211 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.212 28M / 1G INFO General (simplification.cpp : 362) PROCEDURE == Simplification cycle, iteration 12
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Low coverage edge remover
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Low coverage edge remover triggered 0 times
0:02:14.212 28M / 1G INFO StageManager (stage.cpp : 132) STAGE == Simplification Cleanup
0:02:14.212 28M / 1G INFO General (simplification.cpp : 196) PROCEDURE == Post simplification
0:02:14.212 28M / 1G INFO General (graph_simplification.hpp : 453) Disconnection of relatively low covered edges disabled
0:02:14.212 28M / 1G INFO General (graph_simplification.hpp : 489) Complex tip clipping disabled
0:02:14.212 28M / 1G INFO General (graph_simplification.hpp : 634) Creating parallel br instance
0:02:14.212 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.213 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.213 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.223 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.223 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Tip clipper
0:02:14.224 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Tip clipper triggered 0 times
0:02:14.224 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Bulge remover
0:02:14.234 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Bulge remover triggered 0 times
0:02:14.234 28M / 1G INFO General (simplification.cpp : 330) Disrupting self-conjugate edges
0:02:14.235 28M / 1G INFO Simplification (parallel_processing.hpp : 165) Running Removing isolated edges
0:02:14.235 28M / 1G INFO Simplification (parallel_processing.hpp : 167) Removing isolated edges triggered 0 times
0:02:14.235 28M / 1G INFO General (simplification.cpp : 470) Counting average coverage
0:02:14.236 28M / 1G INFO General (simplification.cpp : 476) Average coverage = 58.246
0:02:14.236 28M / 1G INFO StageManager (stage.cpp : 132) STAGE == Contig Output
0:02:14.236 28M / 1G INFO General (contig_output_stage.cpp : 40) Writing GFA to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/assembly_graph_with_scaffolds.gfa
0:02:14.279 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/before_rr.fasta
0:02:14.370 28M / 1G INFO General (contig_output_stage.cpp : 51) Outputting FastG graph to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/assembly_graph.fastg
0:02:14.627 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/simplified_contigs.fasta
0:02:14.720 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/final_contigs.fasta
0:02:14.852 28M / 1G INFO StageManager (stage.cpp : 132) STAGE == Contig Output
0:02:14.852 28M / 1G INFO General (contig_output_stage.cpp : 40) Writing GFA to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/assembly_graph_with_scaffolds.gfa
0:02:14.897 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/before_rr.fasta
0:02:14.992 28M / 1G INFO General (contig_output_stage.cpp : 51) Outputting FastG graph to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/assembly_graph.fastg
0:02:15.250 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/simplified_contigs.fasta
0:02:15.344 28M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K21/final_contigs.fasta
0:02:15.478 28M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:15.492 8M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 15 seconds
Max read length detected as 151
== Running assembler: K33
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K33/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K33/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/dataset.info) with K=33
0:00:00.000 4M / 4M INFO General (main.cpp : 90) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO General (launch.hpp : 51) SPAdes started
0:02:05.760 16M / 1G INFO StageManager (stage.cpp : 132) STAGE == Contig Output
0:02:05.760 16M / 1G INFO General (contig_output_stage.cpp : 40) Writing GFA to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/assembly_graph_with_scaffolds.gfa
0:02:05.800 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/before_rr.fasta
0:02:05.885 16M / 1G INFO General (contig_output_stage.cpp : 51) Outputting FastG graph to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/assembly_graph.fastg
0:02:06.130 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/simplified_contigs.fasta
0:02:06.218 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/final_contigs.fasta
0:02:06.347 16M / 1G INFO StageManager (stage.cpp : 132) STAGE == Contig Output
0:02:06.347 16M / 1G INFO General (contig_output_stage.cpp : 40) Writing GFA to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/assembly_graph_with_scaffolds.gfa
0:02:06.390 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/before_rr.fasta
0:02:06.480 16M / 1G INFO General (contig_output_stage.cpp : 51) Outputting FastG graph to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/assembly_graph.fastg
0:02:06.735 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/simplified_contigs.fasta
0:02:06.826 16M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K33/final_contigs.fasta
0:02:06.958 16M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:06.964 8M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 6 seconds
== Running assembler: K55
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K55/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K55/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/..
..
0:02:31.939 232M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K55/final_contigs.fasta
0:02:32.066 232M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:32.418 12M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 32 seconds
== Running assembler: K77
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K77/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K77/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/dataset.info) with K=77
0:00:00.000 4M / 4M INFO General (main.cpp : 90) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO General (launch.hpp : 51) SPAdes started
0:00:00.000 4M / 4M INFO General (launch.hpp : 58) Starting from stage: construction
0:00:00.000 4M / 4M INFO General (launch.hpp : 65) Two-step RR enabled: 0
..
0:02:28.441 128M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:28.479 8M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 28 seconds
== Running assembler: K99
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K99/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K99/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/dataset.info) with K=99
0:00:00.000 4M / 4M INFO General (main.cpp : 90) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO General (launch.hpp : 51) SPAdes started
0:00:00.000 4M / 4M INFO General (launch.hpp : 58) Starting from stage: construction
0:00:00.000 4M / 4M INFO General (launch.hpp : 65) Two-step RR enabled: 0
0:00:00.001 4M / 4M INFO General (launch.hpp : 76) Will need read mapping, kmer mapper will be attached
0:00:00.001 4M / 4M INFO StageManager (stage.cpp : 132) STAGE == de Bruijn graph construction
..
0:02:21.316 140M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K99/simplified_contigs.fasta
0:02:21.402 140M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K99/final_contigs.fasta
0:02:21.531 140M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:21.588 8M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 21 seconds
== Running assembler: K127
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K127/configs/config.info
0:00:00.000 4M / 4M INFO General (main.cpp : 74) Loaded config from /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/K127/configs/careful_mode.info
0:00:00.000 4M / 4M INFO General (memory_limit.cpp : 49) Memory limit set to 40 Gb
0:00:00.000 4M / 4M INFO General (main.cpp : 87) Starting SPAdes, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 88) Maximum k-mer length: 128
0:00:00.000 4M / 4M INFO General (main.cpp : 89) Assembling dataset (/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/dataset.info) with K=127
0:00:00.000 4M / 4M INFO General (main.cpp : 90) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO General (launch.hpp : 51) SPAdes started
0:00:00.000 4M / 4M INFO General (launch.hpp : 58) Starting from stage: construction
0:00:00.000 4M / 4M INFO General (launch.hpp : 65) Two-step RR enabled: 0
0:00:00.001 4M / 4M INFO General (launch.hpp : 76) Will need read mapping, kmer mapper will be attached
0:00:00.001 4M / 4M INFO StageManager (stage.cpp : 132) STAGE == de Bruijn graph construction
0:00:00.003 4M / 4M INFO General (read_converter.hpp : 59) Binary reads detected
0:00:00.004 4M / 4M INFO General (construction.cpp : 111) Max read length 151
0:00:00.004 4M / 4M INFO General (construction.cpp : 117) Average read length 150.933
0:00:00.004 4M / 4M INFO General (stage.cpp : 101) PROCEDURE == k+1-mer counting
..
0:02:14.702 144M / 1G INFO General (contig_output.hpp : 22) Outputting contigs to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K127/before_rr.fasta
0:02:14.789 144M / 1G INFO General (contig_output_stage.cpp : 51) Outputting FastG graph to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K127/assembly_graph.fastg
0:02:15.033 144M / 1G INFO General (contig_output_stage.cpp : 20) Outputting FastG paths to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K127/final_contigs.paths
0:02:15.154 144M / 1G INFO General (contig_output_stage.cpp : 20) Outputting FastG paths to /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly//K127/scaffolds.paths
0:02:15.311 144M / 1G INFO General (launch.hpp : 149) SPAdes finished
0:02:15.354 8M / 1G INFO General (main.cpp : 109) Assembling time: 0 hours 2 minutes 15 seconds
===== Assembling finished. Used k-mer sizes: 21, 33, 55, 77, 99, 127
===== Mismatch correction started.
== Processing of contigs
== Running contig polishing tool: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-corrector-core /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/contigs/configs/corrector.info /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta
== Dataset description file was created: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/contigs/configs/corrector.info
/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/contigs/configs/log.properties 0:00:00.000 4M / 4M INFO General (main.cpp : 58) Starting MismatchCorrector, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 59) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 195) Splitting assembly...
0:00:00.000 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 196) Assembly file: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta
0:00:00.629 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 203) Processing paired sublib of number 0
0:00:00.629 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 206) /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
0:00:00.630 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 140) Running bwa index ...: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa index -a is /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta
[bwa_index] Pack FASTA... 0.05 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 1.11 seconds elapse.
[bwa_index] Update BWT... 0.03 sec
[bwa_index] Pack forward-only FASTA... 0.01 sec
[bwa_index] Construct SA from BWT and Occ... 0.48 sec
[main] Version: 0.7.12-r1039
[main] CMD: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa index -a is /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta
[main] Real time: 1.816 sec; CPU: 1.689 sec
0:00:02.491 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 149) Running bwa mem ...:/hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa mem -v 1 -t 1 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz > /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/tmp/corrector_u3yc490y/lib0_qEblzI/tmp.sam
[main] Version: 0.7.12-r1039
[main] CMD: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa mem -v 1 -t 1 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_contigs.fasta /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
[main] Real time: 99.016 sec; CPU: 106.835 sec
0:01:41.517 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 209) Adding samfile /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/tmp/corrector_u3yc490y/lib0_qEblzI/tmp.sam
0:01:52.274 48M / 48M INFO DatasetProcessor (dataset_processor.cpp : 105) processed 1000000reads, flushing
0:02:03.447 48M / 48M INFO DatasetProcessor (dataset_processor.cpp : 105) processed 2000000reads, flushing
0:02:04.545 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 235) Processing contigs
0:02:10.606 36M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_1_length_276305_cov_10.791692 processed with 2 changes in thread 0
0:02:15.477 36M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_2_length_243149_cov_10.143588 processed with 0 changes in thread 0
0:02:19.350 32M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_3_length_197455_cov_10.080455 processed with 0 changes in thread 0
0:02:22.948 28M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_4_length_183139_cov_10.101064 processed with 0 changes in thread 0
0:02:26.215 24M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_5_length_165258_cov_10.112747 processed with 0 changes in thread 0
0:02:28.932 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_6_length_138875_cov_10.026631 processed with 0 changes in thread 0
0:02:31.608 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_7_length_135159_cov_10.135857 processed with 0 changes in thread 0
0:02:34.209 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_8_length_132800_cov_10.037302 processed with 0 changes in thread 0
0:02:36.800 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_9_length_132384_cov_10.035091 processed with 0 changes in thread 0
0:02:39.396 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_10_length_132014_cov_10.112847 processed with 0 changes in thread 0
0:02:41.920 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_11_length_128079_cov_10.032606 processed with 0 changes in thread 0
0:02:44.124 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_12_length_111557_cov_10.147160 processed with 0 changes in thread 0
0:02:46.230 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_13_length_105858_cov_10.213192 processed with 0 changes in thread 0
0:02:48.244 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_14_length_102135_cov_10.138920 processed with 0 changes in thread 0
0:02:49.972 16M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_15_length_88537_cov_10.042631 processed with 0 changes in thread 0
0:03:19.537 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_39_length_41772_cov_10.041662 processed with 0 changes in thread 0
0:03:20.339 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_40_length_40483_cov_10.150263 processed with 0 changes in thread 0
0:03:21.126 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_41_length_39822_cov_10.155889 processed with 0 changes in thread 0
0:03:21.856 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_42_length_37235_cov_10.057966 processed with 0 changes in thread 0
0:03:22.564 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_43_length_36180_cov_10.036557 processed with 0 changes in thread 0
0:03:23.198 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_44_length_32026_cov_10.125991 processed with 0 changes in thread 0
0:03:23.809 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_45_length_31111_cov_10.086980 processed with 0 changes in thread 0
0:03:24.409 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_46_length_30878_cov_9.944132 processed with 0 changes in thread 0
0:03:25.010 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_47_length_30822_cov_10.001662 processed with 0 changes in thread 0
0:03:25.598 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_48_length_29723_cov_10.141033 processed with 0 changes in thread 0
0:03:26.113 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_49_length_26457_cov_9.975997 processed with 0 changes in thread 0
0:03:26.524 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_50_length_20861_cov_9.972412 processed with 0 changes in thread 0
0:03:34.831 4M / 48M INFO DatasetProcessor (dataset_processor.cpp : 255) Gluing processed contigs
0:03:34.941 4M / 48M INFO General (main.cpp : 72) Correcting time: 0 hours 3 minutes 34 seconds
== Processing of scaffolds
== Running contig polishing tool: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-corrector-core /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/scaffolds/configs/corrector.info /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta
== Dataset description file was created: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/scaffolds/configs/corrector.info
/hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/mismatch_corrector/scaffolds/configs/log.properties 0:00:00.000 4M / 4M INFO General (main.cpp : 58) Starting MismatchCorrector, built from refs/heads/spades_3.13.0, git revision 8ea46659e9b2aca35444a808db550ac333006f8b
0:00:00.000 4M / 4M INFO General (main.cpp : 59) Maximum # of threads to use (adjusted due to OMP capabilities): 1
0:00:00.000 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 195) Splitting assembly...
0:00:00.000 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 196) Assembly file: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta
0:00:00.614 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 203) Processing paired sublib of number 0
0:00:00.614 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 206) /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
0:00:00.615 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 140) Running bwa index ...: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa index -a is /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta
[bwa_index] Pack FASTA... 0.05 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 1.11 seconds elapse.
[bwa_index] Update BWT... 0.03 sec
[bwa_index] Pack forward-only FASTA... 0.03 sec
[bwa_index] Construct SA from BWT and Occ... 0.48 sec
[main] Version: 0.7.12-r1039
[main] CMD: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa index -a is /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta
[main] Real time: 1.834 sec; CPU: 1.707 sec
0:00:02.459 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 149) Running bwa mem ...:/hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa mem -v 1 -t 1 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz > /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/tmp/corrector_ljrgi6px/lib0_hKba9g/tmp.sam
[main] Version: 0.7.12-r1039
[main] CMD: /hpc/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/share/spades-3.13.0-0/bin/spades-bwa mem -v 1 -t 1 /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/misc/assembled_scaffolds.fasta /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_1_paired.fastq.gz /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/trimmed/GCA_011404755.1_ASM1140475v1_genomic_2_paired.fastq.gz
[main] Real time: 99.052 sec; CPU: 106.864 sec
0:01:41.519 4M / 4M INFO DatasetProcessor (dataset_processor.cpp : 209) Adding samfile /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/tmp/corrector_ljrgi6px/lib0_hKba9g/tmp.sam
0:01:52.248 48M / 48M INFO DatasetProcessor (dataset_processor.cpp : 105) processed 1000000reads, flushing
0:02:03.381 48M / 48M INFO DatasetProcessor (dataset_processor.cpp : 105) processed 2000000reads, flushing
0:02:04.470 4M / 48M INFO DatasetProcessor (dataset_processor.cpp : 235) Processing contigs
0:02:10.441 36M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_1_length_276305_cov_10.791692 processed with 2 changes in thread 0
0:02:15.361 36M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_2_length_243149_cov_10.143588 processed with 0 changes in thread 0
0:02:19.346 32M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_3_length_197455_cov_10.080455 processed with 0 changes in thread 0
0:02:23.038 28M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_4_length_183139_cov_10.101064 processed with 0 changes in thread 0
0:02:26.373 24M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_5_length_165258_cov_10.112747 processed with 0 changes in thread 0
0:02:29.179 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_6_length_139437_cov_10.042115 processed with 5 changes in thread 0
0:02:31.954 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_7_length_138875_cov_10.026631 processed with 0 changes in thread 0
0:02:34.687 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_8_length_135159_cov_10.135857 processed with 0 changes in thread 0
0:02:37.327 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_9_length_132800_cov_10.037302 processed with 0 changes in thread 0
0:02:39.966 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_10_length_132014_cov_10.112847 processed with 0 changes in thread 0
0:02:42.510 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_11_length_128079_cov_10.032606 processed with 0 changes in thread 0
0:02:44.750 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_12_length_111557_cov_10.147160 processed with 0 changes in thread 0
0:02:46.888 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_13_length_105858_cov_10.213192 processed with 0 changes in thread 0
0:02:48.939 20M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_14_length_102135_cov_10.138920 processed with 0 changes in thread 0
0:02:50.693 16M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_15_length_88537_cov_10.042631 processed with 0 changes in thread 0
0:02:52.445 16M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_16_length_87789_cov_10.071525 processed with 0 changes in thread 0
0:03:23.850 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_43_length_36180_cov_10.036557 processed with 0 changes in thread 0
0:03:24.496 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_44_length_32026_cov_10.125991 processed with 0 changes in thread 0
0:03:25.117 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_45_length_31111_cov_10.086980 processed with 0 changes in thread 0
0:03:25.728 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_46_length_30878_cov_9.944132 processed with 0 changes in thread 0
0:03:26.339 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_47_length_30822_cov_10.001662 processed with 0 changes in thread 0
0:03:26.935 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_48_length_29723_cov_10.141033 processed with 0 changes in thread 0
0:03:27.458 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_49_length_26457_cov_9.975997 processed with 0 changes in thread 0
0:03:27.874 8M / 48M INFO DatasetProcessor (dataset_processor.cpp : 251) Contig NODE_50_length_20861_cov_9.972412 processed with 0 changes in thread 0
0:03:36.169 4M / 48M INFO DatasetProcessor (dataset_processor.cpp : 255) Gluing processed contigs
0:03:36.280 4M / 48M INFO General (main.cpp : 72) Correcting time: 0 hours 3 minutes 36 seconds
===== Mismatch correction finished.
- Corrected reads are in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/corrected/
- Assembled contigs are in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/contigs.fasta
- Assembled scaffolds are in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/scaffolds.fasta
- Assembly graph is in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/assembly_graph.fastg
- Assembly graph in GFA format is in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/assembly_graph_with_scaffolds.gfa
- Paths in the assembly graph corresponding to the contigs are in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/contigs.paths
- Paths in the assembly graph corresponding to the scaffolds are in /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/scaffolds.paths
======= SPAdes pipeline finished.
SPAdes log can be found here: /hpc/dla_mm/jpaganini/data/recovering_ecoli_plasmids/testing_plasmidid/20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/spades.log
Thank you for using SPAdes!
Sun Aug 30 14:40:09 CEST 2020
DONE. Assembled contigs can be found at 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/contigs.fasta:
DONE. Assembled scaffolds can be found at 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/assembly/scaffolds.fasta:
Removing unnecesary folders
DONE removing unwanted folders
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/mash_screener.sh
DEPENDENCY STATUS
bash �[0;32mINSTALLED�[0m
mash �[0;32mINSTALLED�[0m
Output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer
creating sketch of 2020-08-19_plasmids.fasta
Sketching plasmidid_db/2020-08-19_plasmids.fasta...
Writing to 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.msh...
Sun Aug 30 14:43:22 CEST 2020
screening reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq
Loading 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.msh...
8782236 distinct hashes.
Streaming from reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq...
Estimated distinct k-mers in pool: 15336880
Summing shared...
Reallocating to winners...
Computing coverage medians...
Writing output...
Sun Aug 30 14:45:36 CEST 2020
DONE Screening GCA_011404755.1_ASM1140475v1_genomic of NO_GROUP Group
Retrieving sequences matching more than 0.95 identity
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/filter_fasta.sh
Output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer
Sun Aug 30 14:45:37 CEST 2020
Filtering terms on file 2020-08-19_plasmids.fasta
Sun Aug 30 14:45:57 CEST 2020
DONE Filtering terms on file 2020-08-19_plasmids.fasta
File with filtered sequences can be found in 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.fasta
Previous number of sequences= 22845
Post number of sequences= 28
Namespace(distance=0.5, input_file='20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.fasta', output=False, output_grouped=False)
Obtaining mash distance
Obtaining cluster from distance
Calculating length
Filtering representative fasta
�[35m28 sequences clustered into 28�[0m
DONE
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/bowtie_mapper.sh
DEPENDENCY STATUS
bowtie2-build �[0;32mINSTALLED�[0m
bowtie2 �[0;32mINSTALLED�[0m
Output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/mapping
Building index of database.filtered_0.95_term.0.5.representative.fasta
Settings:
Output files: "20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta..bt2"
Line rate: 6 (line is 64 bytes)
Lines per side: 1 (side is 64 bytes)
Offset rate: 1 (one in 2)
FTable chars: 10
Strings: unpacked
Max bucket size: default
Max bucket size, sqrt multiplier: default
Max bucket size, len divisor: 4
Difference-cover sample period: 1024
Endianness: little
Actual local endianness: little
Sanity checking: disabled
Assertions: disabled
Random seed: 0
Sizeofs: void:8, int:4, long:8, size_t:8
Input files DNA, FASTA:
20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta
Building a SMALL index
Reading reference sizes
Time reading reference sizes: 00:00:00
Calculating joined length
Writing header
Reserving space for joined string
Joining reference sequences
Time to join reference sequences: 00:00:00
bmax according to bmaxDivN setting: 363379
Using parameters --bmax 272535 --dcv 1024
Doing ahead-of-time memory usage test
Passed! Constructing with these parameters: --bmax 272535 --dcv 1024
Constructing suffix-array element generator
Building DifferenceCoverSample
Building sPrime
Building sPrimeOrder
V-Sorting samples
V-Sorting samples time: 00:00:00
Allocating rank array
Ranking v-sort output
Ranking v-sort output time: 00:00:00
Invoking Larsson-Sadakane on ranks
Invoking Larsson-Sadakane on ranks time: 00:00:00
Sanity-checking and returning
Building samples
Reserving space for 12 sample suffixes
Generating random suffixes
QSorting 12 sample offsets, eliminating duplicates
QSorting sample offsets, eliminating duplicates time: 00:00:00
Multikey QSorting 12 samples
(Using difference cover)
Multikey QSorting samples time: 00:00:00
Calculating bucket sizes
Splitting and merging
Splitting and merging time: 00:00:00
Avg bucket size: 1.45352e+06 (target: 272534)
Converting suffix-array elements to index image
Allocating ftab, absorbFtab
Entering Ebwt loop
Getting block 1 of 1
No samples; assembling all-inclusive block
Sorting block of length 1453518 for bucket 1
(Using difference cover)
Sorting block time: 00:00:00
Returning block of 1453519 for bucket 1
Exited Ebwt loop
fchr[A]: 0
fchr[C]: 358023
fchr[G]: 719961
fchr[T]: 1094992
fchr[$]: 1453518
Exiting Ebwt::buildToDisk()
Returning from initFromVector
Wrote 4681688 bytes to primary EBWT file: 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta.1.bt2
Wrote 2907044 bytes to secondary EBWT file: 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta.2.bt2
Re-opening _in1 and _in2 as input streams
Returning from Ebwt constructor
Headers:
len: 1453518
bwtLen: 1453519
sz: 363380
bwtSz: 363380
lineRate: 6
offRate: 1
offMask: 0xfffffffe
ftabChars: 10
eftabLen: 20
eftabSz: 80
ftabLen: 1048577
ftabSz: 4194308
offsLen: 726760
offsSz: 2907040
lineSz: 64
sideSz: 64
sideBwtSz: 48
sideBwtLen: 192
numSides: 7571
numLines: 7571
ebwtTotLen: 484544
ebwtTotSz: 484544
color: 0
reverse: 0
Total time for call to driver() for forward index: 00:00:00
Reading reference sizes
Time reading reference sizes: 00:00:00
Calculating joined length
Writing header
Reserving space for joined string
Joining reference sequences
Time to join reference sequences: 00:00:00
Time to reverse reference sequence: 00:00:00
bmax according to bmaxDivN setting: 363379
Using parameters --bmax 272535 --dcv 1024
Doing ahead-of-time memory usage test
Passed! Constructing with these parameters: --bmax 272535 --dcv 1024
Constructing suffix-array element generator
Building DifferenceCoverSample
Building sPrime
Building sPrimeOrder
V-Sorting samples
V-Sorting samples time: 00:00:00
Allocating rank array
Ranking v-sort output
Ranking v-sort output time: 00:00:00
Invoking Larsson-Sadakane on ranks
Invoking Larsson-Sadakane on ranks time: 00:00:00
Sanity-checking and returning
Building samples
Reserving space for 12 sample suffixes
Generating random suffixes
QSorting 12 sample offsets, eliminating duplicates
QSorting sample offsets, eliminating duplicates time: 00:00:00
Multikey QSorting 12 samples
(Using difference cover)
Multikey QSorting samples time: 00:00:00
Calculating bucket sizes
Splitting and merging
Splitting and merging time: 00:00:00
Avg bucket size: 1.45352e+06 (target: 272534)
Converting suffix-array elements to index image
Allocating ftab, absorbFtab
Entering Ebwt loop
Getting block 1 of 1
No samples; assembling all-inclusive block
Sorting block of length 1453518 for bucket 1
(Using difference cover)
Sorting block time: 00:00:01
Returning block of 1453519 for bucket 1
Exited Ebwt loop
fchr[A]: 0
fchr[C]: 358023
fchr[G]: 719961
fchr[T]: 1094992
fchr[$]: 1453518
Exiting Ebwt::buildToDisk()
Returning from initFromVector
Wrote 4681688 bytes to primary EBWT file: 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta.rev.1.bt2
Wrote 2907044 bytes to secondary EBWT file: 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/kmer/database.filtered_0.95_term.0.5.representative.fasta.rev.2.bt2
Re-opening _in1 and _in2 as input streams
Returning from Ebwt constructor
Headers:
len: 1453518
bwtLen: 1453519
sz: 363380
bwtSz: 363380
lineRate: 6
offRate: 1
offMask: 0xfffffffe
ftabChars: 10
eftabLen: 20
eftabSz: 80
ftabLen: 1048577
ftabSz: 4194308
offsLen: 726760
offsSz: 2907040
lineSz: 64
sideSz: 64
sideBwtSz: 48
sideBwtLen: 192
numSides: 7571
numLines: 7571
ebwtTotLen: 484544
ebwtTotSz: 484544
color: 0
reverse: 1
Total time for backward call to driver() for mirror index: 00:00:01
Sun Aug 30 14:46:10 CEST 2020
mapping reads/GCA_011404755.1_ASM1140475v1_genomic_R1.fq
mapping reads/GCA_011404755.1_ASM1140475v1_genomic_R2.fq
1000000 reads; of these:
1000000 (100.00%) were paired; of these:
748800 (74.88%) aligned concordantly 0 times
210899 (21.09%) aligned concordantly exactly 1 time
40301 (4.03%) aligned concordantly >1 times
----
748800 pairs aligned concordantly 0 times; of these:
444 (0.06%) aligned discordantly 1 time
----
748356 pairs aligned 0 times concordantly or discordantly; of these:
1496712 mates make up the pairs; of these:
1485688 (99.26%) aligned 0 times
4421 (0.30%) aligned exactly 1 time
6603 (0.44%) aligned >1 times
25.72% overall alignment rate
Sun Aug 30 14:51:16 CEST 2020
DONE Mapping GCA_011404755.1_ASM1140475v1_genomic of NO_GROUP Group
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/sam_to_bam.sh
DEPENDENCY STATUS
samtools �[0;32mINSTALLED�[0m
Default output directory is 20200830_test_output/NO_GROUP/GCA_011404755.1_ASM1140475v1_genomic/mapping
Sun Aug 30 14:51:16 CEST 2020
Converting SAM to sorted indexed BAM in GCA_011404755
Sun Aug 30 14:52:04 CEST 2020
Sorting BAM file in GCA_011404755
Sun Aug 30 14:52:52 CEST 2020
Indexing BAM file in GCA_011404755
Sun Aug 30 14:52:56 CEST 2020
DONE Converting SAM to sorted indexed BAM in GCA_011404755
GCA_011404755.bam removed
#Executing /home/dla_mm/jpaganini/data/miniconda3/envs/plasmid_id/bin/get_coverage.sh
GCA_011404755.1_ASM1140475v1_genomic.sorted.bam not supplied, please, introduce a valid file
ERROR: 1 missing files, aborting execution