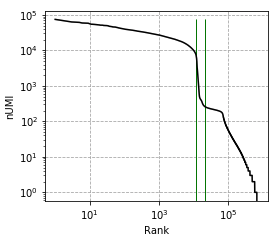
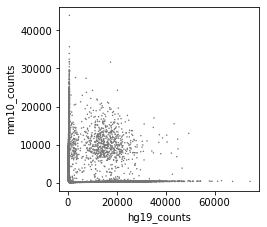
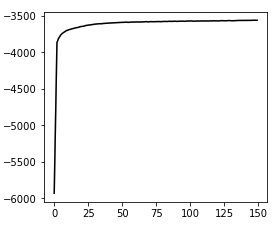
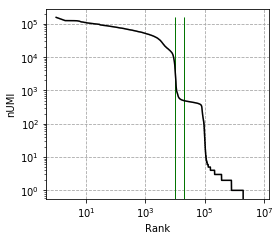
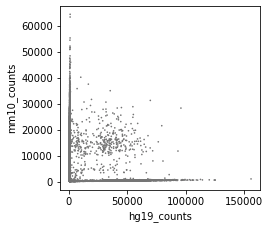
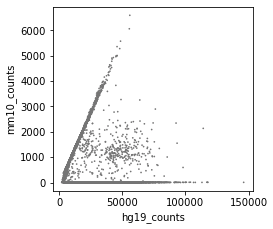
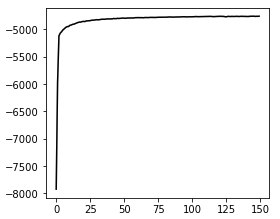
Great package! I am currently using cellbender V2.1. I ran into an issue, which is caused by too high memory allocation.
[....]
cellbender:remove-background: [epoch 198] average training loss: 1790.0774
cellbender:remove-background: [epoch 199] average training loss: 1787.5904
cellbender:remove-background: [epoch 200] average training loss: 1792.2732
cellbender:remove-background: [epoch 200] average test loss: 1773.5361
cellbender:remove-background: Inference procedure complete.
cellbender:remove-background: 2020-08-06 23:06:51
cellbender:remove-background: Preparing to write outputs to file...
cell counts tensor([ 8096., 6134., 1805., 2324., 5410., 5546., 5092., 1724., 5301.,
1329., 3143., 5382., 618., 3833., 6279., 5066., 2166., 7982.,
7920., 3160., 3907., 12285., 3919., 7285., 1576., 2011., 1805.,
5842., 2688., 8696., 7202., 7752., 6153., 4572., 2058., 7318.,
3196., 3786., 7375., 2877., 2555., 4179., 1650., 1776., 4262.,
4624., 5314., 5727., 5470., 693., 4088., 2078., 1429., 2127.,
5265., 649., 4733., 9864., 19365., 7845., 5621., 699., 3006.,
3918., 1308., 6071., 5948., 1816., 7495., 3055., 2016., 11080.,
1845., 1077., 14801., 8278., 2293., 1718., 1436., 7260., 1655.,
13636., 8505., 1307., 2211., 7010., 4465., 1496., 3346., 8285.,
1948., 1978., 2007., 1693., 16839., 6170., 4675., 12212., 1955.,
1499.], device='cuda:0')
Traceback (most recent call last):
File "path/to/bin/cellbender", line 33, in <module>
sys.exit(load_entry_point('cellbender', 'console_scripts', 'cellbender')())
File "path/to/CellBender/cellbender/base_cli.py", line 101, in main
cli_dict[args.tool].run(args)
File "path/to/cellbender/remove_background/cli.py", line 103, in run
main(args)
File "path/to/cellbender/remove_background/cli.py", line 196, in main
run_remove_background(args)
File "path/to/cellbender/remove_background/cli.py", line 166, in run_remove_background
save_plots=True)
File "path/to/cellbender/remove_background/data/dataset.py", line 524, in save_to_output_file
inferred_count_matrix = self.posterior.mean
File "path/to/cellbender/remove_background/infer.py", line 56, in mean
self._get_mean()
File "path/to/lib/python3.7/site-packages/torch/autograd/grad_mode.py", line 15, in decorate_context
return func(*args, **kwargs)
File "path/to/cellbender/remove_background/infer.py", line 402, in _get_mean
alpha_est=map_est['alpha'])
File "path/to/lib/python3.7/site-packages/torch/autograd/grad_mode.py", line 15, in decorate_context
return func(*args, **kwargs)
File "path/to/cellbender/remove_background/infer.py", line 1005, in _lambda_binary_search_given_fpr
alpha_est=alpha_est)
File "path/to/lib/python3.7/site-packages/torch/autograd/grad_mode.py", line 15, in decorate_context
return func(*args, **kwargs)
File "path/to/cellbender/remove_background/infer.py", line 809, in _calculate_expected_fpr_given_lambda_mult
alpha_est=alpha_est)
File "path/to/lib/python3.7/site-packages/torch/autograd/grad_mode.py", line 15, in decorate_context
return func(*args, **kwargs)
File "path/to/cellbender/remove_background/infer.py", line 604, in _true_counts_from_params
.log_prob(noise_count_tensor)
File path/to/lib/python3.7/site-packages/torch/distributions/poisson.py", line 63, in log_prob
return (rate.log() * value) - rate - (value + 1).lgamma()
RuntimeError: CUDA out of memory. Tried to allocate 1016.00 MiB (GPU 0; 3.97 GiB total capacity; 2.48 GiB already allocated; 378.79 MiB free; 2.58 GiB reserved in total by PyTorch)
Do you suggest to change environmental settings, or adjust the batch size? Changing "empty-drop-training-fraction" did not solve the issue. Thanks for your thoughts!