xinglab / rmats-turbo Goto Github PK
View Code? Open in Web Editor NEWLicense: Other
License: Other






















































































































When I run rmats.py, an error appeared as follows:
line 1: 2784173 Segmentation fault (core dumped) , but the core file cannot be generated; I don’t know why; The python version I used is 2.7.13,
the code is
python /software/rmats-turbo/rmats.py --b1 MV411-C.txt --b2 MV411-T.txt --gtf ref_annot.gtf -t paired --readLength 120 --nthread 4 --od ./output --tmp ./tmp_output
Is there anything else that needs to be set or modified? thanks very much .
I am still seeing a lot of stuff from the development and testing of rMATS kind of scattered around:
GLM_MS_unpaired 3.04.01 PM.py
analysis.py
diff_result1.txt
output.txt
rMATS_Result_P.txt
rMATS_Result_P_backup.txt
test.c
test.txt
test_output.txt
trans.exe
From a Q&A standpoint it's now possible (thanks to the open sourcing) to point people to the core statistics behind rMATS e.g. https://www.biostars.org/p/444274/
but it's fairly difficult to isolate the relevant parts of rMATS with all this clutter.
Is it possible to segregate these files in some other directory or branch?
Hi,
I'm using rmats-turbo for splicing events detection and quite new to it. I'm compare 2 samples without replicate. However, for each sample, there are fastq.gz files sequenced from 2 lanes. Do I need to merge reads from multiple lane together before I run rmats or I can simply create the text file like this? Will rmats-turbo use the correct algorithm for the no-replicates samples?
sample_A.txt
sample_A_lane1_R1.fq.gz:sample_A_lane1_R2.fq.gz,sample_A_lane2_R1.fq.gz:sample_A_lane2_R2.fq.gz
sample_B.txt
sample_B_lane1_R1.fq.gz:sample_B_lane1_R2.fq.gz,sample_B_lane2_R1.fq.gz:sample_B_lane2_R2.fq.gz
Best,
JL
my run code like following, to compare two paired-groups A and D:
srun ./software/wrapped_rmats.sh \
./'sheepH_A_bamlist.txt' ./'sheepH_D_bamlist.txt' \ #bam file path of two group
./human.gtf \ #gtf annotation
48 \ #threads
. /outputdir/AS_AD_compare2A \ #outputfile
./temp_dir/tmp_$RANDOM >log_AD 2>&1 & #temp_file randomly named
my output file only has header: [AS type ].MATS.JCEC.txt
ID GeneID geneSymbol chr strand 1stExonStart_0base 1stExonEnd 2ndExonStart_0base 2ndExonEnd upstreamES upstreamEE downstreamES downstreamEE ID IJC_SAMPLE_1 SJC_SAMPLE_1 IJC_SAMPLE_2 SJC_SAMPLE_2 IncFormLen SkipFormLen PValue FDR IncLevel1 IncLevel2 IncLevelDifference
the log file
module add R-3.6.1
gtf: 4.323092699050903
There are 27054 distinct gene ID in the gtf file
There are 29118 distinct transcript ID in the gtf file
There are 25073 one-transcript genes in the gtf file
There are 239130 exons in the gtf file
There are 6070 one-exon transcripts in the gtf file
There are 6070 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 1.076292
Average number of exons per transcript is 8.212446
Average number of exons per transcript excluding one-exon tx is 10.111940
Average number of gene per geneGroup is 4.199280
statistic: 0.010064125061035156
novel: 483.66304659843445
The splicing graph and candidate read have been saved into /public1/home/sc30941/transcription_analysis/ay_sheep/pj1_sheepNative_res/tmp_2738/2020-06-24-17:33:46_053614.rmats
save: 0.006211042404174805
loadsg: 0.001512765884399414
==========
Done processing each gene from dictionary to compile AS events
Found 364 exon skipping events
Found 39 exon MX events
Found 385 alt SS events
There are 329 alt 3 SS events and 56 alt 5 SS events.
Found 216 RI events
==========
ase: 0.25577592849731445
count: 0.25571632385253906
Processing count files.
Done processing count files.
Hi,
I have previously used MATS, it was really essential to some of my previous work. I am looking forward to trying rMATS.
I have tried to install rMATS with conda as instructed. I had initially encountered a few idiosyncratic stumbling blocks with MacOSX, but found workarounds. However, I ultimately encountered the following issue when I try to run ./test_mats:
Collecting package metadata (current_repodata.json): done
Solving environment: done
All requested packages already installed.
test (tests.alternative_3_splice_site_novel.test.NovelJunction) ... FAIL
test (tests.alternative_3_splice_site_novel.test.NovelSpliceSite) ... FAIL
test (tests.alternative_5_splice_site_novel.test.NovelJunction) ... FAIL
test (tests.alternative_5_splice_site_novel.test.NovelSpliceSite) ... FAIL
test (tests.mutually_exclusive_exons_novel.test.NovelJunction) ... FAIL
test (tests.mutually_exclusive_exons_novel.test.NovelSpliceSite) ... FAIL
test (tests.only_one_sample.test.StatOffTest) ... FAIL
test (tests.only_one_sample.test.StatOnTest) ... FAIL
test (tests.paired_stats.test.FilteredEventTest) ... FAIL
test (tests.paired_stats.test.OneEventTest) ... FAIL
test (tests.paired_stats.test.TwoEventTest) ... FAIL
test (tests.prep_post.test.Test) ... FAIL
test (tests.retained_intron_novel.test.NovelJunction) ... FAIL
test (tests.retained_intron_novel.test.NovelSpliceSite) ... FAIL
test (tests.skipped_exon_basic.test.Test) ... FAIL
test (tests.skipped_exon_novel.test.NovelJunction) ... FAIL
test (tests.skipped_exon_novel.test.NovelSpliceSite) ... FAIL
test (tests.variable_read_length.test.Length1Test) ... FAIL
test (tests.variable_read_length.test.Length1VariableTest) ... FAIL
test (tests.variable_read_length.test.Length2Test) ... FAIL
test (tests.variable_read_length.test.Length2VariableTest) ... FAIL
All of the fails are similar, for example:
FAIL: test (tests.alternative_3_splice_site_novel.test.NovelJunction)
Traceback (most recent call last):
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/tests/alternative_3_splice_site_novel/test.py", line 95, in test
self._run_test()
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/tests/base_test.py", line 17, in _run_test
self._check_results()
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/tests/alternative_3_splice_site_novel/test.py", line 114, in _check_results
self._check_no_error_results()
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/tests/base_test.py", line 61, in _check_no_error_results
self.assertEqual(self._rmats_return_code, 0)
AssertionError: 1 != 0
Looking in the tests directory, they all say the following in the stderror:
Traceback (most recent call last):
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmats.py", line 16, in
from rmatspipeline import run_pipe
ImportError: dlopen(/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so, 2): Symbol not found: _GOMP_parallel
Referenced from: /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so
Expected in: flat namespace
in /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so
Test run environment info:
active environment : /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/conda_envs/test_rmats
active env location : /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/conda_envs/test_rmats
shell level : 2
user config file : /Users/nnp/.condarc
populated config files :
conda version : 4.8.4
conda-build version : not installed
python version : 3.8.3.final.0
virtual packages : __osx=10.15.6
base environment : /Users/nnp/opt/miniconda3 (writable)
channel URLs : https://repo.anaconda.com/pkgs/main/osx-64
https://repo.anaconda.com/pkgs/main/noarch
https://repo.anaconda.com/pkgs/r/osx-64
https://repo.anaconda.com/pkgs/r/noarch
package cache : /Users/nnp/opt/miniconda3/pkgs
/Users/nnp/.conda/pkgs
envs directories : /Users/nnp/opt/miniconda3/envs
/Users/nnp/.conda/envs
platform : osx-64
user-agent : conda/4.8.4 requests/2.24.0 CPython/3.8.3 Darwin/19.6.0 OSX/10.15.6
UID:GID : 501:20
netrc file : None
offline mode : False
If it helps, even when I run ./run_rmats -h
Traceback (most recent call last):
File "/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmats.py", line 16, in
from rmatspipeline import run_pipe
ImportError: dlopen(/Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so, 2): Symbol not found: _GOMP_parallel
Referenced from: /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so
Expected in: flat namespace
in /Users/nnp/projects/lab/global_software/MATS/rmats-turbo/rmatspipeline.cpython-36m-darwin.so
From my searches, it seems like sometimes this is related to an OpenMP issue in MacOSX. I encountered an issue related to that earlier on, and I did have to use gcc-10 to make the files in rMATS_C. Any ideas what has gone awry?
Hi,
Currently, I want to output all the detected splicing events by changing the "cstat" to zero, so there will be no cutoff for splicing difference. However, the results are little bit werid by changing "cstat".
The following commands both output the same results.
python .../rMATS.4.0.2/rMATS-turbo-Linux-UCS4/rmats.py --b1 ctrl.txt --b2 sh1.txt --od as1_v3 -t paired --readLength 150 --cstat 0.001 --libType fr-unstranded --nthread 16 --gtf .../genomes/rat/star/Rattus_norvegicus.Rnor_6.0.96.chr.gtf
python .../rMATS.4.0.2/rMATS-turbo-Linux-UCS4/rmats.py --b1 ctrl.txt --b2 sh1.txt --od as1_v4 -t paired --readLength 150 --cstat 0 --libType fr-unstranded --nthread 16 --gtf .../genomes/rat/star/Rattus_norvegicus.Rnor_6.0.96.chr.gtf
python .../rMATS.4.0.2/rMATS-turbo-Linux-UCS4/rmats.py --b1 ctrl.txt --b2 sh1.txt --od as1_v5 -t paired --readLength 150 --cstat 0.1 --libType fr-unstranded --nthread 16 --gtf .../genomes/rat/star/Rattus_norvegicus.Rnor_6.0.96.chr.gtf
There are 32623 distinct gene ID in the gtf file
There are 40808 distinct transcript ID in the gtf file
There are 27245 one-transcript genes in the gtf file
There are 310574 exons in the gtf file
There are 9929 one-exon transcripts in the gtf file
There are 9599 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 1.250897
Average number of exons per transcript is 7.610616
Average number of exons per transcript excluding one-exon tx is 9.736229
Average number of gene per geneGroup is 4.035672Done processing each gene from dictionary to compile AS events
Found 11343 exon skipping events
Found 998 exon MX events
Found 688 alt SS events
There are 418 alt 3 SS events and 270 alt 5 SS events.
Found 420 RI eventsRunning the statistical part.
The statistical part is done.
Done.
$cat ctrl.txt
R19021274-ZNY-1.sorted.bam,R19021274-ZNY-4.sorted.bam,R19021274-ZNY-7.sorted.bam,R19021274-ZNY-10.sorted.bam
$cat sh1.txt
#R19021274-ZNY-2.sorted.bam,R19021274-ZNY-5.sorted.bam,R19021274-ZNY-8.sorted.bam,R19021274-ZNY-11.sorted.bam
I don't know if I understand this issue correctly, so any help would be appreciated.
Thanks!
Hi,
Great tool!
I was trying to find out how rMATS is handling paired-end RNAseq data. Is each read treated separately or are the counts somehow combined per fragment?
Hi,
I noticed probably since rMATS version 3.x you started supporting FASTAQ as inputs. I was wondering which aligner does rMATS use under the hood in the latest version? Is it STAR?
I tried looking up this information in your Docs and FAQs, but I was not sure.
Thanks!
Hi~
when I according the protocol to install rmats-turbo. I code ././build_rmats --conda ,it prompt me bash: gsl-config: command not found;But I checked my path,and typed gsl-config work well。 I don't know why it not work。
Preparing transaction: done
Verifying transaction: done
Executing transaction: done
bash: gsl-config: command not found
(base) [zpliu@mn02 rmats-turbo]$ gsl-config
Usage: gsl-config [OPTION]
Known values for OPTION are:
--prefix show GSL installation prefix
--libs print library linking information, with cblas
--libs-without-cblas print library linking information, without cblas
--cflags print pre-processor and compiler flags
--help display this help and exit
--version output version information
An external CBLAS library can be specified using the GSL_CBLAS_LIB
environment variable. The GSL CBLAS library is used by default.Best Regards
Hi,
I have a quick question. Can I use rMATs without having two conditions ? I have large panel of samples and I am interested to calculate PSIs across samples. This could be used for QTL analysis or for identifying co-splicing modules ( like co-expression modules).
Hi,
i have installed rmats but some issues occur.
when i run cmd ./build_rmats --conda, it is done with last line
** using staged installation
** R
** data
*** moving datasets to lazyload DB
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
** building package indices
** testing if installed package can be loaded from temporary location
** testing if installed package can be loaded from final location
** testing if installed package keeps a record of temporary installation path
- DONE (PAIRADISE)
Loading required package: PAIRADISE
and i run python rmats.py
it prints this message
ImportError: /data/tools/rMATS_install/rmats-turbo/rmatspipeline.cpython-37m-x86_64-linux-gnu.so: undefined symbol: _ZNK8BamTools12BamAlignment10GetTagTypeERKSsRc
what can i do?
Best Regards
Jeongmin
I got some trouble when using the pre-built version rmats_turbo_v4_1_0_python_3_6.tar.gz:
python rmats.py -h
Traceback (most recent call last):
File "rmats.py", line 16, in <module>
from rmatspipeline import run_pipe
ModuleNotFoundError: No module named 'rmatspipeline'The code I got rmats_turbo_v4_1_0_python_3_6.tar.gz:
get https://github.com/Xinglab/rmats-turbo/releases/download/v4.1.0/rmats_turbo_v4_1_0_python_3_6.tar.gz
tar -zxvf rmats_turbo_v4_1_0_python_3_6.tar.gzHi,
I am using rMATs to compute the differentially AS events between two groups of RNA-seq data. Regarding to the AS events with statistical FDR values, I got two types of files: [AS_Event].MATS.JC.txt and [AS_Event].MATS.JCEC.txt. These two files differ both in the numbers of detected AS events, as well as the FDR values for the same event. Which file should I use for the downstream analysis?
Additionally, I also plan to detect splicing QTLs, as the matched genomic sequencing data is also available. I would prefer to use the PSI (percent of splicing index) value as the phenotype for each AS event, and my question is where can I get the PSI values from the rMATs output?
Thanks and have a nice day!
Best wishes,
Wenyu Zhang
Hi, rMATS was installed with conda (https://anaconda.org/bioconda/rmats). fastq files were aligned OK but there was no output/data in .MATS.JCEC.txt files. fromGTF..txt files were OK. Any idea what went wrong? Thanks!
Hi,
I used rmats-turbo with threads more than 1 to my research, it came out to be crashed and threw out a Segmentation fault, but when i set the threads number to be 1, it ran smoothly, how did it happened, can you figure it out? Btw, it also come to the old released version, like 4.0.2. The error information listed as below:
$ rmats.py --gtf final.gtf --b1 B366_45.txt --b2 D366_45.txt -t paired --libType fr-unstranded --readLength 150 \
--nthread 2 --od B366-45-1_B366-45-2_B366-45-3_vs_D366-45-1_D366-45-2_D366-45-3_1 \
--novelSS --mil 20 --mel 1000 --tmp 366_45_tmp --paired-stats
gtf: 2.76681184769
There are 109280 distinct gene ID in the gtf file
There are 142631 distinct transcript ID in the gtf file
There are 88364 one-transcript genes in the gtf file
There are 907449 exons in the gtf file
There are 17267 one-exon transcripts in the gtf file
There are 16423 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 1.305189
Average number of exons per transcript is 6.362214
Average number of exons per transcript excluding one-exon tx is 7.100779
Average number of gene per geneGroup is 1.378398
statistic: 0.0358090400696
Segmentation fault (core dumped)
Dear,
According to rMATS document, --od contains the final output files from the post step:
[AS_Event].MATS.JC.txt: Final output including only reads that span junctions defined by rmats (Junction Counts)
[AS_Event].MATS.JCEC.txt: Final output including both reads that span junctions defined by rmats (Junction Counts) and reads that do not cross an exon boundary (Exon Counts)
.....
There are so many outputs that I am confused, which file should I use to identify the significant alternative splicing events ? There were no replicates in my experiment, so are the FDR and p-value still meaningful ?
I am installing rmats on a cluster and I am having an issue with the installation.
I have load following modules
1) blas/3.8.0 2) gcc/7.3.0 3) lapack/3.9.0 4) gsl/2.5-cjj 5) cmake/3.15.5
but when run ./build_rmats warings and errors occurs
I have tried to test different gcc versions(like 7.3 6.4 8.3 etc,but not 5.4 which version is not installed by root ),all failed
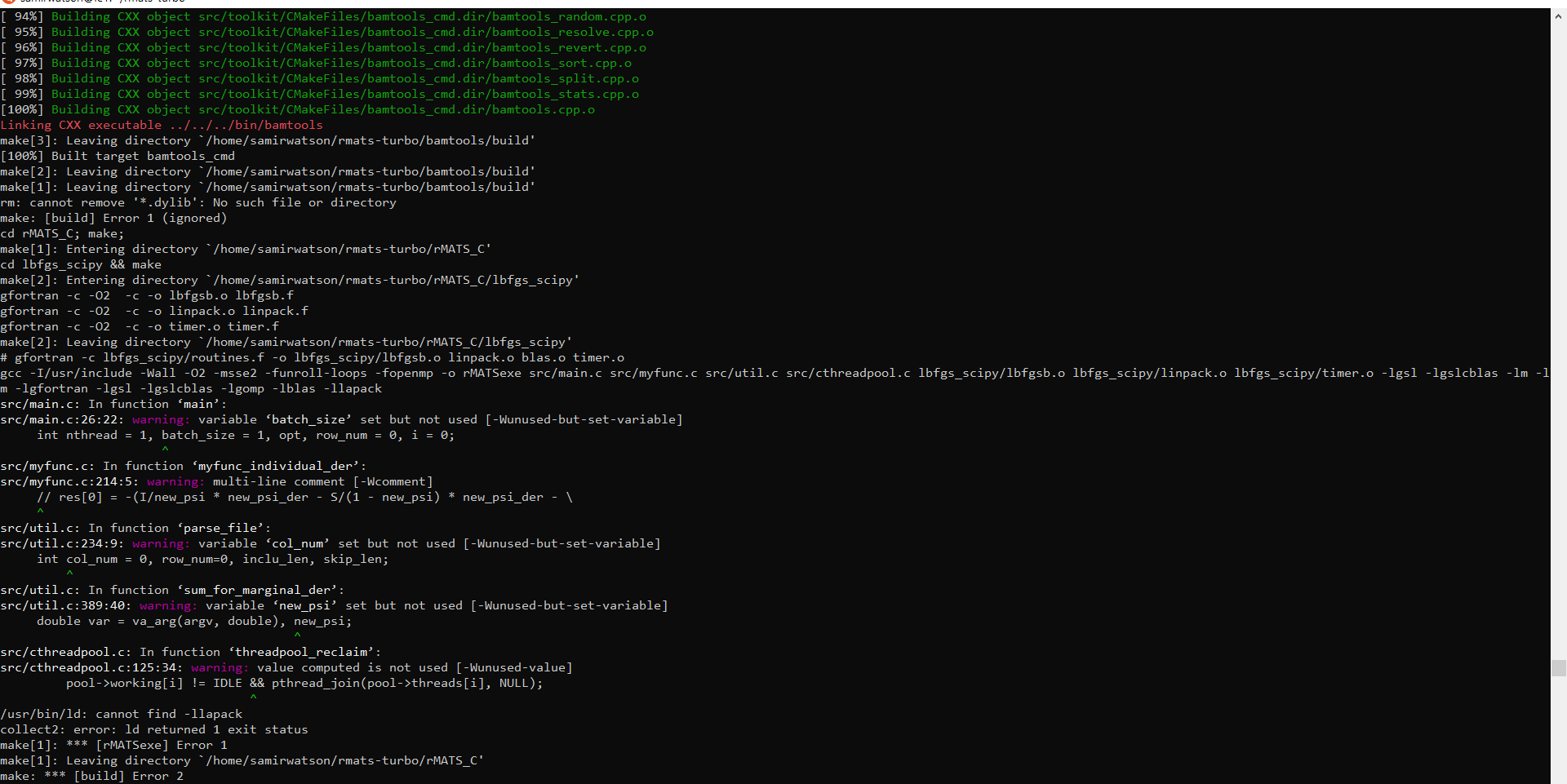
I capture the first errors and log the proccess.
("C:\Users\Administrator\Documents\github_err_rmats.png")
using GCC 8.3:
log_compile_rmats.txt
I try to reorganize the output:
warning:
1. warning: comparison between signed and unsigned integer expressions [-Wsign-compare]
if ( numBytesWritten != (sizeof(offsetCount) + linearOffsets.size()*sizeof(uint64_t)) )
2. rmats-turbo/bamtools/src/api/internal/io/HostAddress_p.cpp:327:17: note: ...this statement, but the latter is misleadingly indented as if it is guarded by the ‘if’
ss << hex << ( (uint16_t(m_ip6Address[2*i]) << 8) |
^~
3. rmats-turbo/bamtools/src/api/internal/io/HostAddress_p.cpp:325:13: warning: this ‘if’ clause does not guard... [-Wmisleading-indentation]
if ( i != 0 )
^~
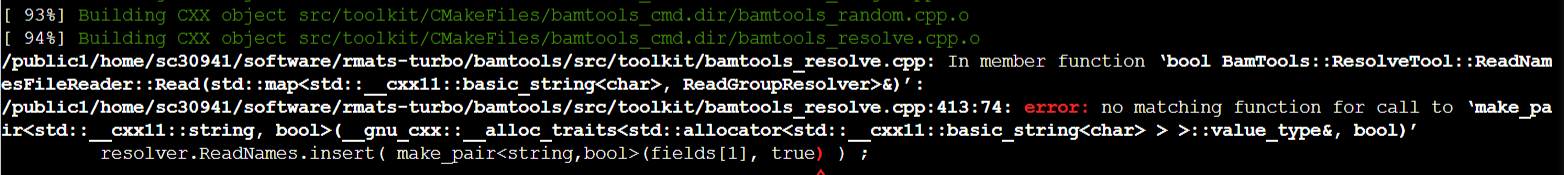
4. rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:413:74: error: no matching function for call to ‘make_pair(__gnu_cxx::__alloc_traits<std::allocator<std::__cxx11::basic_string<char> > >::value_type&, bool)’
resolver.ReadNames.insert( make_pair<string,bool>(fields[1], true) ) ;
^
5. gcc-6.4.0/include/c++/6.4.0/bits/stl_pair.h:497:5: note: candidate: template<class _T1, class _T2> constexpr std::pair<typename std::__decay_and_strip<_Tp>::__type, typename std::__decay_and_strip<_T2>::__type> std::make_pair(_T1&&, _T2&&)
make_pair(_T1&& __x, _T2&& __y)
6. gcc-6.4.0/include/c++/6.4.0/bits/stl_pair.h:497:5: note: template argument deduction/substitution failed:
7. rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:610:75: error: no matching function for call to ‘make_pair(const string&, ReadGroupResolver&)’
readGroups.insert( make_pair<string, ReadGroupResolver>(name, resolver) );
^
8. rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:610:61: note: cannot convert ‘name’ (type ‘const string {aka const std::__cxx11::basic_string<char>}’) to type ‘std::__cxx11::basic_string<char>&&’
readGroups.insert( make_pair<string, ReadGroupResolver>(name, resolver) );
^~~~
9. rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1017:93: error: no matching function for call to ‘make_pair(std::__cxx11::string&, const bool&)’
else resolver.ReadNames.insert( make_pair<string, bool>(al.Name, isCurrentMateUnique) );
^
10. rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1049:93: error: no matching function for call to ‘make_pair(const string&, ReadGroupResolver)’
m_readGroups.insert( make_pair<string, ReadGroupResolver>(rg.ID, ReadGroupResolver()) );
^
11.
make[3]: *** [src/toolkit/CMakeFiles/bamtools_cmd.dir/bamtools_resolve.cpp.o] Error 1
make[2]: *** [src/toolkit/CMakeFiles/bamtools_cmd.dir/all] Error 2
make[1]: *** [all] Error 2
rm: cannot remove `*.dylib': No such file or directory
make: [build] Error 1 (ignored)
src/main.c: In function ‘main’:
src/main.c:26:22: warning: variable ‘batch_size’ set but not used [-Wunused-but-set-variable]
int nthread = 1, batch_size = 1, opt, row_num = 0, i = 0;
^~~~~~~~~~
src/myfunc.c: In function ‘myfunc_individual_der’:
src/myfunc.c:214:5: warning: multi-line comment [-Wcomment]
// res[0] = -(I/new_psi * new_psi_der - S/(1 - new_psi) * new_psi_der - \
^
src/util.c: In function ‘parse_file’:
src/util.c:234:9: warning: variable ‘col_num’ set but not used [-Wunused-but-set-variable]
int col_num = 0, row_num=0, inclu_len, skip_len;
^~~~~~~
src/util.c: In function ‘sum_for_marginal_der’:
src/util.c:389:40: warning: variable ‘new_psi’ set but not used [-Wunused-but-set-variable]
double var = va_arg(argv, double), new_psi;
^~~~~~~
src/cthreadpool.c: In function ‘threadpool_reclaim’:
src/cthreadpool.c:125:34: warning: value computed is not used [-Wunused-value]
pool->working[i] != IDLE && pthread_join(pool->threads[i], NULL);
^~
/vol6/software/libraries/lapack/3.8.0/lib64/../lib64/libblas.a(dcopy.f.o): In function `dcopy_':
/vol-th/home/zhenggang/project/lapack/lapack-3.8.0-intel2013-vol6/BLAS/SRC/dcopy.f:(.text+0xbe): undefined reference to `_intel_fast_memcpy'
collect2: error: ld returned 1 exit status
make[1]: *** [rMATSexe] Error 1
make: *** [build] Error 2
similar warnings do not display
As the title shows, I tried two data sets and multiple attempts. Events with an FDR less than 0.05 are very rare。
the Rmats code runs as follows
python /media/Extend_1/malu2019/miniconda3/bin/rmats.py --b1 bam_name.csv --b2 other_bam_name.csv --gtf /media/Extend_1/malu2019/reference/hg19_ensemble/ Homo_sapiens.GRCh37.87.gtf --od output -t single --nthread 8 --tmp tmp_output --readLength 38 --variable-read-length
the STAR code runs as follows
cat ../../stroma_SRR.csv|while read line;do STAR --runThreadN 20 --genomeDir /media/Extend_1/malu2019/reference/hg19_ensemble/STAR_hg19_refence --outFileNamePrefix ${line} --readFilesIn ${line}_1.fastq.gz ${line}_2.fastq.gz --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outBAMsortingThreadN 10 --alignEndsType EndToEnd; done
Where do you think the error might have occurred?
Thank you very much for your reply
I was able to get rMATS up and running via a conda installation. I used this link:
https://anaconda.org/bioconda/rmats
And it states the current version is 4.1.0. However, in my download, it says the conda package is rmats-4.0.2 and when I use rmats.py --version I get v.3.1.0.
I assume the conda package number doesn't correspond to the rMATS package number? Or perhaps I am doing something wrong. If there is there a conda install for v4.1.0, I would appreciate any tips you have on obtaining it. If there isn't, could we get one?
The output of rmats=4.1.0 with only header, pls check, thank you.
rmats.py --b1 b1.txt --b2 b2.txt --gtf ./gene.gtf --od ../rmats --tmp ./tmp
wc -l ../rmats/*
1 A3SS.MATS.JCEC.txt
1 A3SS.MATS.JC.txt
1 A5SS.MATS.JCEC.txt
1 A5SS.MATS.JC.txt
29 fromGTF.A3SS.txt
14 fromGTF.A5SS.txt
14 fromGTF.MXE.txt
...
Hi!
I met ERROR when ./build_rmats, could someone help me about this? I have already installed the required dependencies. The error is :


Thanks for your help!
Hi, I have some confused when I run the rmats. I now try to find different splicing events between two different sex plant samples.
1、when I used the rmats3.2.5 , if I run the data using the paired stat model, then I will got about five thousands differents events and the most popuplar different splicing event is the RI, which was reports to be the most popular splice type in plant. Then, because some of my sample only have two replicates, so I needed to work with the unparied stastic model, I got fewer splicing events (about 1 thousand , fdr <0.01) and the most poplar splicing events is the SE type. So I do not know, why?
2、 I also try the rmats4, but I got no results, although these data worked when I used the rmats3.2.5.
paired stat model
F M
A3SS | 1090 | 1002
A5SS | 635 | 419
SE | 567 | 351
RI | 2588 | 2301
MXE | 60 | 62
unpaired stat model
F M
A3SS | 250 | 149
A5SS | 212 | 75
SE | 922 | 630
RI | 225 | 88
MXE | 8 | 8
Hi again!
In the experiment I am trying to apply rMATS to, I have 2 conditions, genotype and diet. When I divided the samples by just one condition (genotype), everything worked fine, apart from few differential splicing events :)
However, when I tried to separate the samples by two conditions, the script gave me the following error:
/opt/sge/default/spool/hpc10/job_scripts/62788: line 13: 3224 Segmentation fault (core dumped) /home/ashumskiy/anaconda3/bin/python /home/ashumskiy/Programs/rmats-turbo/rmats.py --b1 /home/ashumskiy/SIRT6/rMATS/WTND.txt --b2 /home/ashumskiy/SIRT6/rMATS/KOND.txt --gtf /home/ashumskiy/Mus_musculus.GRCm38.100.gtf -t single --readLength 90 --nthread 4 --od /home/ashumskiy/SIRT6/rMATS/WTND_vs_KOND --tmp /home/ashumskiy/SIRT6/rMATS/tmp --variable-read-length
/opt/sge/default/spool/hpc10/job_scripts/62788: line 18: 3255 Segmentation fault (core dumped) /home/ashumskiy/anaconda3/bin/python /home/ashumskiy/Programs/rmats-turbo/rmats.py --b1 /home/ashumskiy/SIRT6/rMATS/WTND.txt --b2 /home/ashumskiy/SIRT6/rMATS/KOHFD.txt --gtf /home/ashumskiy/Mus_musculus.GRCm38.100.gtf -t single --readLength 90 --nthread 4 --od /home/ashumskiy/SIRT6/rMATS/WTND_vs_KOHFD --tmp /home/ashumskiy/SIRT6/rMATS/tmp --variable-read-length
/opt/sge/default/spool/hpc10/job_scripts/62788: line 23: 3275 Segmentation fault (core dumped)
/home/ashumskiy/anaconda3/bin/python /home/ashumskiy/Programs/rmats-turbo/rmats.py --b1 /home/ashumskiy/SIRT6/rMATS/WTND.txt --b2 /home/ashumskiy/SIRT6/rMATS/WTHFD.txt --gtf /home/ashumskiy/Mus_musculus.GRCm38.100.gtf -t single --readLength 90 --nthread 4 --od /home/ashumskiy/SIRT6/rMATS/WTND_vs_WTHFD --tmp /home/ashumskiy/SIRT6/rMATS/tmp --variable-read-length
Traceback (most recent call last):
File "rmatspipeline/rmatspipeline.pyx", line 2929, in rmats.rmatspipeline._load_job
ValueError: '/home/ashumskiy/SIRT6/results_star/bam/mouse-25122---B1.bam' is not in list
Exception ignored in: 'rmats.rmatspipeline._load_job'
The last part with the Python error happened a couple of times, with the mouse-25122, as well as another one, all of which are contained in the three runs that suffered a segmentation fault. All other runs went smoothly.
I am definitely sure the path is right and the files are not corrupted, would be grateful for some help in this matter!
Hello,
I have run rMATS turbo v4.1.0 and I was wondering where can I find the p-values (and their FDR adjustments) for the novel splice-site predictions? As far as I can tell, the only predictions that have p-values are those in *.MATS.JC.txt which do not include the IDs of the novel splice-site predictions found in fromGTF.novelSpliceSite.*.txt.
Side question: on your wiki you state:
Reads can be mapped independently of rMATS with any aligner and then the resulting BAM files can be used as input to rMATS.
However on your FAQ you mention:
Q: Can I run rMATS with STAR aligner output?
A: STAR aligner performs soft clipping by default which will generate variable read lengths. You can run STAR with "--alignEndsType EndToEnd" option to suppress soft clipping.
So which one of the two is it? Can rMATS handle any aligner, or special care needs to be paid to soft-clipping. If that's the case shouldn't it be mentioned in bold somewhere? I have already mapped upwards to 100 samples with STAR soft-clipping on. Can I just pass the --variable-read-length flag to rMATS and hope for the best?
Thank you!
Hi,
Let's us directly go into the main points.
I used the command as below:
/build_rmats --conda
The last returned information showed as below:
Preparing transaction: done
Verifying transaction: done
Executing transaction: done
Loading required package: nloptr
Loading required package: foreach
Loading required package: doParallel
Loading required package: iterators
Loading required package: parallel
Loading required package: PAIRADISE
But! When I run:
./run_rmats
The error came to my screen:
Traceback (most recent call last):
File "/home/cmq/software/rmats-turbo/rmats.py", line 16, in
from rmatspipeline import run_pipe
ImportError: /home/cmq/software/rmats-turbo/rmatspipeline.cpython-36m-x86_64-linux-gnu.so: undefined symbol: ZNK8BamTools12BamAlignment14SetErrorStringERKNSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEES8
I activated the environment in building step and then ran again, But the error still exists.
Do you have any idea can fix this problem.
Since the rMATs can be installed by conda, I tried to skip the building step and directly analyzed my data.
My command to install packages: conda install -c bioconda rmats. (python 2.7)
I ran the command:
gtf=/home/yhw/genome/ensembl/release97/mus_musculus/dna_anno/Mus_musculus.GRCm38.97.gtf
rmats.py --b1 dox_bamfile.txt --b2 nodox_bamfile.txt -t paired --od ${OutDir} --gtf ${gtf} --tmp ./temp --readLength 123 --variable-read-length --nthread 12 --tstat 6 --cstat 0.05 --libType fr-unstranded > ${log_file} 2>&1
my bam txt files (dox_bamfile.txt)
./results/star/no_rmrRNA_unique/SRR4032346_1_val_1_SRR4032346_2_val_2Aligned.sortedByCoord.out.bam,./results/star/no_rmrRNA_unique/SRR4032347_1_val_1_SRR4032347_2_val_2Aligned.sortedByCoord.out.bam
(nodox_bamfile.txt)
./results/star/no_rmrRNA_unique/SRR4032348_1_val_1_SRR4032348_2_val_2Aligned.sortedByCoord.out.bam,./results/star/no_rmrRNA_unique/SRR4032349_1_val_1_SRR4032349_2_val_2Aligned.sortedByCoord.out.bam
I have checked the consistency of chromosome name between bam file and gtf file. My genome fa file and gtf file were downloaded from ENSEMBL. STAR was used to align sequences to genome.
The returned information:
gtf: 15.9409089088
There are 55573 distinct gene ID in the gtf file
There are 142333 distinct transcript ID in the gtf file
There are 34470 one-transcript genes in the gtf file
There are 839112 exons in the gtf file
There are 26884 one-exon transcripts in the gtf file
There are 21787 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 2.561190
Average number of exons per transcript is 5.895414
Average number of exons per transcript excluding one-exon tx is 7.035384
Average number of gene per geneGroup is 7.477396
statistic: 0.0188231468201
novel: 275.361290932
The splicing graph and candidate read have been saved into ./temp/2020-08-05-10:17:36_394168.rmats
save: 0.000603199005127
loadsg: 0.000283002853394
==========
Done processing each gene from dictionary to compile AS events
Found 18864 exon skipping events
Found 728 exon MX events
Found 8323 alt SS events
There are 5310 alt 3 SS events and 3013 alt 5 SS events.
Found 4082 RI events
==========
ase: 1.25338506699
count: 0.19796705246
Processing count files.
Done processing count files.
But! the results are empty !!!
After adding parameter --statoff, I got the non-empty results. However, I want to find the differential splicing events. Do you have any suggestions to figure it out?
Just now, I change the --cstat as 0.5 and ran again.
gtf=/home/yhw/genome/ensembl/release97/mus_musculus/dna_anno/Mus_musculus.GRCm38.97.gtf
rmats.py --b1 dox_bamfile.txt --b2 nodox_bamfile.txt -t paired --od ${OutDir} --gtf ${gtf} --tmp ./temp --readLength 123 --variable-read-length --nthread 12 --tstat 6 --cstat 0.5 --libType fr-unstranded > ${log_file} 2>&1
The results named with MATS are still empty~
I don't believe all differential splicing events are non-significanyly. I guess the package wasn't installed correctly. Hence, I'm tring to re-installed the packages without using conda.
Your timely reply and precious advices can save my life. It took me two days to fix these problems.
Hanwen Yu
I am trying to build rmats_turbo_v4_1_0 either the original or the python3.6 version (what is the difference by the way?) and I get the following compilation error:
[ 86%] Building CXX object src/toolkit/CMakeFiles/bamtools_cmd.dir/bamtools_resolve.cpp.o
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp: In member function ‘bool BamTools::ResolveTool::ReadNamesFileReader::Read(std::map, ReadGroupResolver>&)’:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:413:74: error: no matching function for call to ‘make_pair(__gnu_cxx::__alloc_traits > >::value_type&, bool)’
resolver.ReadNames.insert( make_pair(fields[1], true) ) ;
^
In file included from /usr/include/c++/7/bits/stl_algobase.h:64:0,
from /usr/include/c++/7/bits/char_traits.h:39,
from /usr/include/c++/7/string:40,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_tool.h:14,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.h:13,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:10:
/usr/include/c++/7/bits/stl_pair.h:524:5: note: candidate: template constexpr std::pair::__type, typename std::__decay_and_strip<_T2>::__type> std::make_pair(_T1&&, _T2&&)
make_pair(_T1&& __x, _T2&& __y)
^~~~~~~~~
/usr/include/c++/7/bits/stl_pair.h:524:5: note: template argument deduction/substitution failed:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:413:74: note: cannot convert ‘fields.std::vector >::operator[](1)’ (type ‘__gnu_cxx::__alloc_traits > >::value_type {aka std::__cxx11::basic_string}’) to type ‘std::__cxx11::basic_string&&’
resolver.ReadNames.insert( make_pair(fields[1], true) ) ;
^
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp: In member function ‘bool BamTools::ResolveTool::StatsFileReader::ParseReadGroupLine(const string&, std::map, ReadGroupResolver>&)’:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:610:75: error: no matching function for call to ‘make_pair(const string&, ReadGroupResolver&)’
readGroups.insert( make_pair(name, resolver) );
^
In file included from /usr/include/c++/7/bits/stl_algobase.h:64:0,
from /usr/include/c++/7/bits/char_traits.h:39,
from /usr/include/c++/7/string:40,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_tool.h:14,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.h:13,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:10:
/usr/include/c++/7/bits/stl_pair.h:524:5: note: candidate: template constexpr std::pair::__type, typename std::__decay_and_strip<_T2>::__type> std::make_pair(_T1&&, _T2&&)
make_pair(_T1&& __x, _T2&& __y)
^~~~~~~~~
/usr/include/c++/7/bits/stl_pair.h:524:5: note: template argument deduction/substitution failed:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:610:61: note: cannot convert ‘name’ (type ‘const string {aka const std::__cxx11::basic_string}’) to type ‘std::__cxx11::basic_string&&’
readGroups.insert( make_pair(name, resolver) );
^~~~
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp: In member function ‘bool BamTools::ResolveTool::ResolveToolPrivate::MakeStats()’:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1017:93: error: no matching function for call to ‘make_pair(std::__cxx11::string&, const bool&)’
else resolver.ReadNames.insert( make_pair(al.Name, isCurrentMateUnique) );
^
In file included from /usr/include/c++/7/bits/stl_algobase.h:64:0,
from /usr/include/c++/7/bits/char_traits.h:39,
from /usr/include/c++/7/string:40,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_tool.h:14,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.h:13,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:10:
/usr/include/c++/7/bits/stl_pair.h:524:5: note: candidate: template constexpr std::pair::__type, typename std::__decay_and_strip<_T2>::__type> std::make_pair(_T1&&, _T2&&)
make_pair(_T1&& __x, _T2&& __y)
^~~~~~~~~
/usr/include/c++/7/bits/stl_pair.h:524:5: note: template argument deduction/substitution failed:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1017:68: note: cannot convert ‘al.BamTools::BamAlignment::Name’ (type ‘std::__cxx11::string {aka std::__cxx11::basic_string}’) to type ‘std::__cxx11::basic_string&&’
else resolver.ReadNames.insert( make_pair(al.Name, isCurrentMateUnique) );
~~~^~~~
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp: In member function ‘void BamTools::ResolveTool::ResolveToolPrivate::ParseHeader(const BamTools::SamHeader&)’:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1049:93: error: no matching function for call to ‘make_pair(const string&, ReadGroupResolver)’
m_readGroups.insert( make_pair(rg.ID, ReadGroupResolver()) );
^
In file included from /usr/include/c++/7/bits/stl_algobase.h:64:0,
from /usr/include/c++/7/bits/char_traits.h:39,
from /usr/include/c++/7/string:40,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_tool.h:14,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.h:13,
from /opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:10:
/usr/include/c++/7/bits/stl_pair.h:524:5: note: candidate: template constexpr std::pair::__type, typename std::__decay_and_strip<_T2>::__type> std::make_pair(_T1&&, _T2&&)
make_pair(_T1&& __x, _T2&& __y)
^~~~~~~~~
/usr/include/c++/7/bits/stl_pair.h:524:5: note: template argument deduction/substitution failed:
/opt/rmats-turbo/bamtools/src/toolkit/bamtools_resolve.cpp:1049:70: note: cannot convert ‘rg.BamTools::SamReadGroup::ID’ (type ‘const string {aka const std::__cxx11::basic_string}’) to type ‘std::__cxx11::basic_string&&’
m_readGroups.insert( make_pair(rg.ID, ReadGroupResolver()) );
~~~^~
src/toolkit/CMakeFiles/bamtools_cmd.dir/build.make:254: recipe for target 'src/toolkit/CMakeFiles/bamtools_cmd.dir/bamtools_resolve.cpp.o' failed
make[3]: *** [src/toolkit/CMakeFiles/bamtools_cmd.dir/bamtools_resolve.cpp.o] Error 1
Hi All,
I am trying to run the paired analysis. It seems like its very very slow. After few hours (5-6hours), when I checked the tail pairadise_status.txt shows ExonID 3191.[1% completed].
I did a "pre" step to get all ".rmats" file and I am running post step with --paired-stats --nthread 12 on a cluster node. This is on 66 paired samples.
I previously ran a rMATS on 400 samples with --statoff and it ran quite quickly.
Is PAIRADISE known to be slow or something wrong with my data ?
The contents of outdir:
-rw------- 1 gatla rds-000259 31902870 Jun 6 13:09 fromGTF.SE.txt
-rw------- 1 gatla rds-000259 26610924 Jun 6 13:09 fromGTF.novelJunction.SE.txt
-rw------- 1 gatla rds-000259 104 Jun 6 13:09 fromGTF.novelSpliceSite.SE.txt
-rw------- 1 gatla rds-000259 12451609 Jun 6 13:09 fromGTF.MXE.txt
-rw------- 1 gatla rds-000259 11841177 Jun 6 13:09 fromGTF.novelJunction.MXE.txt
-rw------- 1 gatla rds-000259 140 Jun 6 13:09 fromGTF.novelSpliceSite.MXE.txt
-rw------- 1 gatla rds-000259 3947067 Jun 6 13:09 fromGTF.A3SS.txt
-rw------- 1 gatla rds-000259 1640099 Jun 6 13:09 fromGTF.novelJunction.A3SS.txt
-rw------- 1 gatla rds-000259 102 Jun 6 13:09 fromGTF.novelSpliceSite.A3SS.txt
-rw------- 1 gatla rds-000259 3540331 Jun 6 13:09 fromGTF.A5SS.txt
-rw------- 1 gatla rds-000259 1680842 Jun 6 13:09 fromGTF.novelJunction.A5SS.txt
-rw------- 1 gatla rds-000259 102 Jun 6 13:09 fromGTF.novelSpliceSite.A5SS.txt
-rw------- 1 gatla rds-000259 2655352 Jun 6 13:09 fromGTF.RI.txt
-rw------- 1 gatla rds-000259 210140 Jun 6 13:09 fromGTF.novelJunction.RI.txt
-rw------- 1 gatla rds-000259 108 Jun 6 13:09 fromGTF.novelSpliceSite.RI.txt
-rw------- 1 gatla rds-000259 225347532 Jun 6 13:21 JCEC.raw.input.SE.txt
-rw------- 1 gatla rds-000259 83087728 Jun 6 13:21 JC.raw.input.MXE.txt
-rw------- 1 gatla rds-000259 90264130 Jun 6 13:21 JCEC.raw.input.MXE.txt
-rw------- 1 gatla rds-000259 28368148 Jun 6 13:21 JC.raw.input.A3SS.txt
-rw------- 1 gatla rds-000259 29403603 Jun 6 13:21 JCEC.raw.input.A3SS.txt
-rw------- 1 gatla rds-000259 24792986 Jun 6 13:21 JC.raw.input.A5SS.txt
-rw------- 1 gatla rds-000259 26021270 Jun 6 13:21 JCEC.raw.input.A5SS.txt
-rw------- 1 gatla rds-000259 20597476 Jun 6 13:21 JC.raw.input.RI.txt
-rw------- 1 gatla rds-000259 21839709 Jun 6 13:21 JCEC.raw.input.RI.txt
-rw------- 1 gatla rds-000259 213332219 Jun 6 13:22 JC.raw.input.SE.txt
The contents of tmp dirr:
JC_SE/
|-- rMATS_result_.txt
|-- rMATS_result_I-L.txt
|-- rMATS_result_ID.txt
|-- rMATS_result_INP.txt
`-- rMATS_result_paired.txt
tail rMATS_result_paired.txt
Loading required package: nloptr
Loading required package: doParallel
Loading required package: foreach
Loading required package: iterators
Loading required package: parallel
Loading data...
A total of 316481 exons will be tested.
Preparing 1 clusters for parallel processing....
Starting analysis.
It it took to reach 3000 exons in 5-6 hours, I am sure it will never finish for 316481 exons.
Hi,
it seems the tools is not reacting to the C-c signal.
I notice when trying to kill the process after receiving a error message for missing files.
[Edit1] Eventually it listened to the signal, but it was about a minute later.
[Edit2] According to traceback it received the signal at the print statement on line 3072, just after the call to detect_novel(). Maybe it is possible to listen to kill signal in this function?
gtf: 3.531550407409668
There are 46904 distinct gene ID in the gtf file
There are 61451 distinct transcript ID in the gtf file
There are 40212 one-transcript genes in the gtf file
There are 273641 exons in the gtf file
There are 26343 one-exon transcripts in the gtf file
There are 25328 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 1.310144
Average number of exons per transcript is 4.452995
Average number of exons per transcript excluding one-exon tx is 7.043922
Average number of gene per geneGroup is 5.651775
statistic: 0.004889249801635742
^CTraceback (most recent call last):
File "/home/agosdsc/projects/GASSER_mrg1_rnaseq/conda-env/bin/rmats.py", line 431, in <module>
main()
File "/home/agosdsc/projects/GASSER_mrg1_rnaseq/conda-env/bin/rmats.py", line 406, in main
run_pipe(args)
File "rmatspipeline/rmatspipeline.pyx", line 3072, in rmats.rmatspipeline.run_pipe
KeyboardInterrupt
Best,
Alex
I am running following command for a single sample as it is mentioned here: http://rnaseq-mats.sourceforge.net/rmats3.2.4/faq.html
python rMATS.4.0.3beta/rMATS-turbo-Linux-UCS4/rmats.py --b1 b1.txt --statoff --gtf Homo_sapiens.GRCh37.75.gtf --od outputDir -t paired --readLength 101 --libType fr-unstranded --nthread 20
b1.txt contains path for one bam file.
I am getting the following error:
Traceback (most recent call last):
File "rMATS.4.0.3beta/rMATS-turbo-Linux-UCS4/rmats.py", line 316, in
main()
File "rMATS.4.0.3beta/rMATS-turbo-Linux-UCS4/rmats.py", line 297, in main
run_stat(jc_it % ('SE'), args.tstat, 'JC', 'SE', args.cstat, args.od, args.tmp, args.stat)
File "rMATS.4.0.3beta/rMATS-turbo-Linux-UCS4/rmats.py", line 235, in run_stat
validate_countfile(istat)
File "rMATS.4.0.3beta/rMATS-turbo-Linux-UCS4/rmats.py", line 204, in validate_countfile
incv2 = map(int, eles[3].split(','))
ValueError: invalid literal for int() with base 10: ''
Hi,
I used rmats v4.1.0, and get successful report. but the JCEC.txt, JC.txt, fromGTF.novelJunction.txt,JCEC.raw.input.txt, JC.raw.input.txt and MATS.JCEC.txt only have a title, without other informations. And i found that files in tmp/JC_A3SS only have a title too, like rMATS_result_FDR.txt.
rmats.py --nthread 20 --b1 ${input}/F02.txt --b2 ${input}/F01.txt --gtf ${gtf} --od ${output}/LT_control -t paired --tmp ${output}/LT_control/tmp --readLength 140 --cstat 0.01
gtf: 2.58114409447
There are 48077 distinct gene ID in the gtf file
There are 91258 distinct transcript ID in the gtf file
There are 31454 one-transcript genes in the gtf file
There are 609665 exons in the gtf file
There are 17352 one-exon transcripts in the gtf file
There are 11996 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 1.898163
Average number of exons per transcript is 6.680675
Average number of exons per transcript excluding one-exon tx is 8.014410
Average number of gene per geneGroup is 5.401415
statistic: 0.00837898254395
novel: 411.806879044
The splicing graph and candidate read have been saved into
tmp/2020-07-30-08:45:53_898
315.rmats
save: 0.0171041488647
loadsg: 0.00025486946106
==========
Done processing each gene from dictionary to compile AS events
Found 1134 exon skipping events
Found 13 exon MX events
Found 4675 alt SS events
There are 3041 alt 3 SS events and 1634 alt 5 SS events.
Found 6468 RI events
==========
ase: 0.623385906219
count: 0.25501203537
Processing count files.
Done processing count files.
Below is an example of a transcript structure that shows differential exon usage between WT and geneX using DexSeq (C. elegans data). I expected this gene/transcripts also to be part of the rmats output. However it does not show up. Upon closer inspection it already does not seem to be part of the fromGTF[rest].txt files? Is that expected behaviour?

Hi Eric,
Sorry, I don't quite understand the results in fromGTF.novelJunction.[AS_Event].txt file. Is this means some novel junctions can be found in my RNA samples but not annotated by GTF file? Or any junctions can be found by my samples in spite of GTF annotation?
Best regards,
Sofia
I'm trying to use rMATS for studying differential splicing in a panel of various RNA-binding protein (RBP) knockdown (KD) experiments. More precisely, I have 20 RBPs, for every RBP I have 2 control replicates and 2 KD replicates (so, 80 bam files in total).
I ran rMATS in --task prep mode, putting all the bam files into the b1.txt file, and using 80 threads. The prep step successfully accomplished.
Then I would like to run a post step for each RBP individually (so, run 20 post steps) but unfortunately rMATS requires all the same bam files to be present in the post step as well.
Is it possible to run the post step on a subset of bam files from the prep step?
Hi!
I am trying to set up rMats. I first ran ./build_rmats and then ran ./test_rmats.
Every test fails. Is the test suite currently unfinished and this is expected, or is something wrong?
Thanks!
Hello,
I'm using rMATs v.4.1.0 for my analysis.
My input is rmats.py --nthread 30 --b1 E4_treated.txt --b2 E4_control.txt --gtf /merged_gtf.annotated_IDmodified.gtf --od E4_result -t paired --variable-read-length --tmp tmp
Howerver I got an python error of division by zero:
==========
Done processing each gene from dictionary to compile AS events
Found 25035 exon skipping events
Found 1291 exon MX events
Found 45822 alt SS events
There are 29779 alt 3 SS events and 16043 alt 5 SS events.
Found 25347 RI events
==========
ase: 4.069897890090942
count: 23.107282400131226
Processing count files.
Traceback (most recent call last):
File "/apps/mats/4.1.0/rMATS/rMATS_P/inclusion_level.py", line 56, in
psi2=vec2psi(inc2,skp2,effective_inclusion_length,effective_skipping_length);
File "/apps/mats/4.1.0/rMATS/rMATS_P/inclusion_level.py", line 30, in vec2psi
psi.append(str(round(float(inc[i])/inclusion_length/(float(inc[i])/inclusion_length+float(skp[i])/skipping_length),3)));
ZeroDivisionError: float division by zero
The bam files are sorted bam output by STAR, and the gtf file is my assembled transcripts result. How can I work around this issue?
Thanks
Ziliang
Hi,
I have use rMATS with the --novelSS option,
I was wonder whether rMATS is able to test all introns for potential retention or it is restricted to those annotated as "retained introns" in the gtf file.
Why some introns are not present in the output file RI.MATS.JC[EC].txt, is that because they do not have enough reads to be quantified or there are other filters applied?
Thank you very much for the help.
With previous versions of rMATS we used rMATS-STAT to estimate p-values. I would like to run rmats-turbo with --statoff and then perform statistics after the rMATS analysis. Can rMATS-STAT still be used? Is there a different method you would recommend?
Hi,
Thanks for the work getting rmats into bioconda.
For now rmats in bioconda sits at version 4.1.0, but for OSX bioconda version is still at 3.2.5.
Are there any plans to have the OSX version updated as well ?
Kind regards,
Warddeb
I have tried all options but none works. Building from master or the release source code gives exact this output with or without the CXXFLAGS option:

I currently only have long reads from ONT, and I want to use rMATS. I was wondering how I could use/modify the parameters to use rMATS with long reads?
Hi,
I have get a summary.txt, but I have a question about it.
I want to know whether the result only calculates the number of inclusion exon, even if it is a SE event.
But in my opinion, SE should calculate the exclusion exon.
So houw to understand it?

I have downloaded: rmats_turbo_v4_1_0.tar.gz extracted and ran ./build_rmats --conda --no-paired-model which completed with a few warnings along the way but no noticeable errors. When I run ./test_rmats it builds the conda env with no warnings/errors but I end up with 21 failures. Do you have any recommendations?
Hi,
We can build it using this tarball, after adding library references for blas and lapack to the C Makefile. But when we try to run "python rmats.py", we get this error:
$ python rmats.py help
Traceback (most recent call last):
File "rmats.py", line 16, in
from rmatspipeline import run_pipe
ImportError: /hpc/software/rMATS_turbo/v4.1.0/rmatspipeline.so: undefined symbol: ZNK8BamTools12BamAlignment14SetErrorStringERKNSt7__cxx1112basic_stringIcSt11char_traitsIcESaIcEEES8
It appears to come from the bamtools source, rather than the pipeline source. How can we fix this?
Hi,
I was wondering if a Bioconda release for 4.1.0 is planned soon ?
Kind regards,
Warddeb
Hi, I am installing rmats on a cluster and I am having an issue with the installation where I get the following error.
Hi!
I successfully used the ./build_rmats, but now that I am trying to run the program itself, I am getting the following error:
Traceback (most recent call last):
File "/home/ashumskiy//Programs/rmats-turbo/rmats.py", line 16, in
from rmatspipeline import run_pipe
ImportError: No module named rmatspipeline
Hello,
I want to run summary.py to generate a summary.txt by this command:
nohup python summary.py output_dir /home/lvjin/rmats-turbo/CGGA/CGGA_test/ --inc-level-diff-cutoff 0.1 --summary-path /home/lvjin/rmats-turbo/CGGA/summary/ --summary-prefix /home/lvjin/rmats-turbo/CGGA/summary/summary &
but it's wrong:
summary.py: error: unrecognized arguments: /home/lvjin/rmats-turbo/CGGA/CGGA_test/
so what can I do?
Thanks!
Hi
Is there an easy way to get the transcripts (from the gtf file) that contain specific rMATS events?
Do you have a tool that I can use out of the box?
Thank you in advance
Best
Foivos
Hi,
We have some paired-end RNA-seq samples. The read length of them before trimming is 150. We trimmed pair-end reads by trim_galore. The reads in the raw fastq files do not have the same length.
We then used STAR to align the trimmed fastqs using --alignEndsType Local (default option with soft-clipping allowed).
My questions are:
(1) Can I run rMATS v4.1.0? With --variable-read-length –readLength 150 (length before trimming)?
(2) If not, should we trim the reads (R1, R2) to the same length (how long) and align them with STAR --alignEndsType EndToEnd?
Thanks and hope for your reply.
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.