

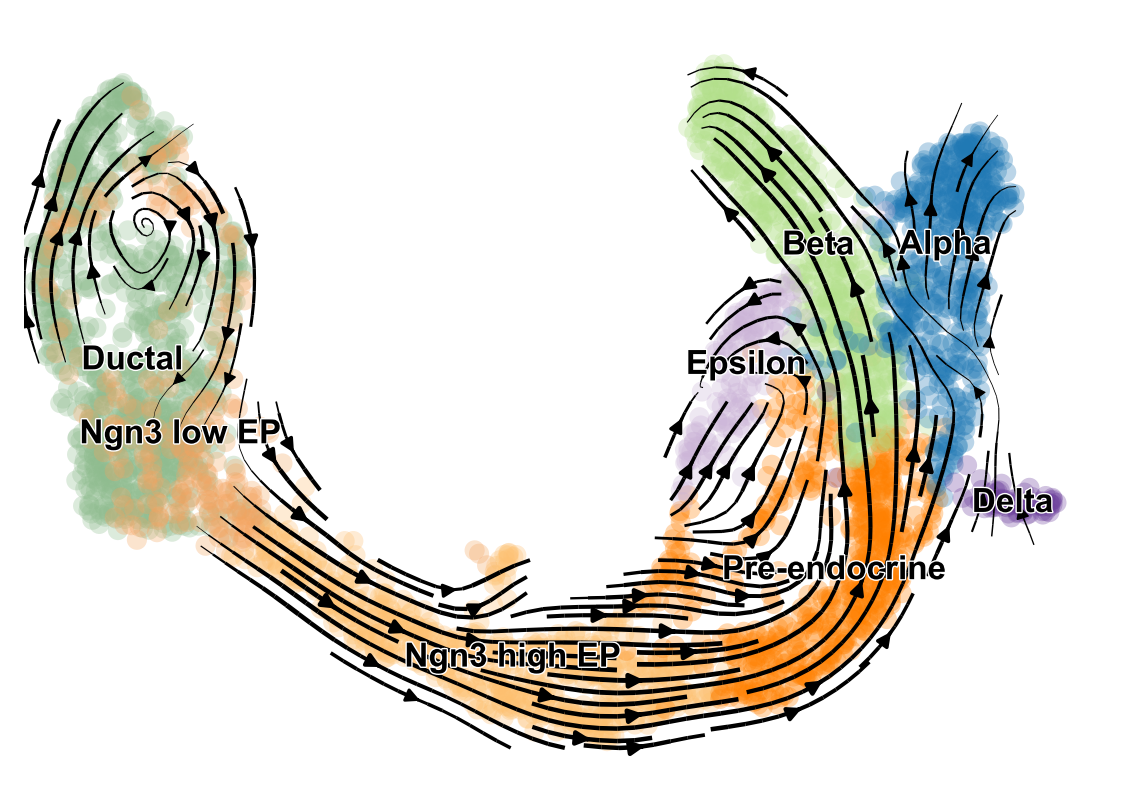
scVelo is a scalable toolkit for RNA velocity analysis in single cells; RNA velocity enables the recovery of directed dynamic information by leveraging splicing kinetics 1. scVelo collects different methods for inferring RNA velocity using an expectation-maximization framework 2, deep generative modeling 3, or metabolically labeled transcripts4.
- estimate RNA velocity to study cellular dynamics.
- identify putative driver genes and regimes of regulatory changes.
- infer a latent time to reconstruct the temporal sequence of transcriptomic events.
- estimate reaction rates of transcription, splicing and degradation.
- use statistical tests, e.g., to detect different kinetics regimes.
If you include or rely on scVelo when publishing research, please adhere to the following citation guide:
If you use the EM (dynamical) or steady-state model, cite
@article{Bergen2020,
title = {Generalizing RNA velocity to transient cell states through dynamical modeling},
volume = {38},
ISSN = {1546-1696},
url = {http://dx.doi.org/10.1038/s41587-020-0591-3},
DOI = {10.1038/s41587-020-0591-3},
number = {12},
journal = {Nature Biotechnology},
publisher = {Springer Science and Business Media LLC},
author = {Bergen, Volker and Lange, Marius and Peidli, Stefan and Wolf, F. Alexander and Theis, Fabian J.},
year = {2020},
month = aug,
pages = {1408–1414}
}If you use veloVI (VI model), cite
@article{Gayoso2023,
title = {Deep generative modeling of transcriptional dynamics for RNA velocity analysis in single cells},
ISSN = {1548-7105},
url = {http://dx.doi.org/10.1038/s41592-023-01994-w},
DOI = {10.1038/s41592-023-01994-w},
journal = {Nature Methods},
publisher = {Springer Science and Business Media LLC},
author = {Gayoso, Adam and Weiler, Philipp and Lotfollahi, Mohammad and Klein, Dominik and Hong, Justin and Streets, Aaron and Theis, Fabian J. and Yosef, Nir},
year = {2023},
month = sep
}If you use the implemented method for estimating RNA velocity from metabolic labeling information, cite
@article{Weiler2023,
title = {Unified fate mapping in multiview single-cell data},
url = {http://dx.doi.org/10.1101/2023.07.19.549685},
DOI = {10.1101/2023.07.19.549685},
publisher = {Cold Spring Harbor Laboratory},
author = {Weiler, Philipp and Lange, Marius and Klein, Michal and Pe’er, Dana and Theis, Fabian J.},
year = {2023},
month = jul
}Found a bug or would like to see a feature implemented? Feel free to submit an issue. Have a question or would like to start a new discussion? Head over to GitHub discussions. Your help to improve scVelo is highly appreciated. For further information visit scvelo.org.































































































































